![]() Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
1. NAAM VAN HET GENEESMIDDEL
Kerendia 10 mg filmomhulde tabletten
Kerendia 20 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Kerendia 10 mg filmomhulde tabletten
Elke filmomhulde tablet bevat 10 mg finerenon.
Hulpstof met bekend effect
Elke filmomhulde tablet bevat 45 mg lactose (als monohydraat); zie rubriek 4.4.
Kerendia 20 mg filmomhulde tabletten
Elke filmomhulde tablet bevat 20 mg finerenon.
Hulpstof met bekend effect
Elke filmomhulde tablet bevat 40 mg lactose (als monohydraat); zie rubriek 4.4.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet)
Kerendia 10 mg filmomhulde tabletten
Roze, langwerpige, ovale filmomhulde tablet met een lengte van 10 mm en een breedte van 5 mm, gemarkeerd met ‘10’ aan één zijde en ‘FI’ aan de andere zijde.
Kerendia 20 mg filmomhulde tabletten
Gele, langwerpige, ovale filmomhulde tablet met een lengte van 10 mm en een breedte van 5 mm, gemarkeerd met ‘20’ aan één zijde en ‘FI’ aan de andere zijde.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Kerendia is geïndiceerd voor de behandeling van chronische nierschade (met albuminurie) bij diabetes mellitus type 2 bij volwassenen.
Voor onderzoeksresultaten met betrekking tot renale en cardiovasculaire voorvallen, zie rubriek 5.1.
4.2 Dosering en wijze van toediening
Dosering
De aanbevolen streefdosis is 20 mg finerenon eenmaal daags.
De aanbevolen maximumdosis is 20 mg finerenon eenmaal daags.
Instellen van de behandeling
Het serumkalium en de geschatte glomerulaire filtratiesnelheid (eGFR) moeten worden gemeten om te bepalen of een behandeling met finerenon kan worden ingesteld en om de startdosis te bepalen.
Als het serumkalium ≤ 4,8 mmol/l is, kan een behandeling met finerenon worden ingesteld. Voor monitoring van het serumkalium, zie ‘Voortzetting van de behandeling’ hieronder.
Als het serumkalium > 4,8 tot 5,0 mmol/l is, kan overwogen worden om de behandeling met finerenon in te stellen met aanvullende monitoring van het serumkalium tijdens de eerste 4 weken op basis van de kenmerken van de patiënt en de serumkaliumwaarden (zie rubriek 4.4).
Als het serumkalium > 5,0 mmol/l is, mag een behandeling met finerenon niet worden ingesteld (zie rubriek 4.4).
De aanbevolen startdosis van finerenon wordt gebaseerd op de eGFR en wordt in tabel 1 weergegeven.
Tabel 1: Instellen van een behandeling met finerenon en aanbevolen dosis
eGFR (ml/min/1,73 m2) | Startdosis (eenmaal daags) |
≥ 60 | 20 mg |
≥ 25, < 60 | 10 mg |
< 25 | Niet aanbevolen |
Voortzetting van de behandeling
Het serumkalium en de eGFR moeten 4 weken na het instellen of het hervatten van de behandeling met finerenon of na een verhoging van de dosis opnieuw worden gemeten (zie tabel 2 voor informatie over voortzetting van een behandeling met finerenon en aanpassing van de dosis).
Daarna moet het serumkalium regelmatig en wanneer de kenmerken van de patiënt en de serumkaliumwaarden dat noodzakelijk maken, opnieuw worden gemeten.
Zie rubriek 4.4 en 4.5 voor meer informatie.
Tabel 2: Voortzetting van de behandeling met finerenon en aanpassing van de dosis
| Huidige dosis finerenon (eenmaal daags) | ||
10 mg | 20 mg | ||
Huidig serumkalium (mmol/l) | ≤ 4,8 | Verhoog tot 20 mg finerenon eenmaal daags* | Handhaaf 20 mg eenmaal daags |
> 4,8 tot 5,5 | Handhaaf 10 mg eenmaal daags | Handhaaf 20 mg eenmaal daags | |
> 5,5 | Onderbreek de behandeling met finerenon. | Onderbreek de behandeling met finerenon. | |
* Handhaaf 10 mg eenmaal daags als de eGFR is verminderd met > 30% ten opzichte van de vorige meting
Vergeten dosis
Een vergeten dosis moet zo snel mogelijk nadat de patiënt het merkt, worden ingenomen maar uitsluitend op dezelfde dag.
De patiënt mag geen 2 doses innemen om een vergeten dosis in te halen.
Speciale populaties
Ouderen
Een dosisaanpassing is niet nodig bij oudere patiënten (zie rubriek 5.2).
Nierfunctiestoornis
Instellen van de behandeling
Bij patiënten met een eGFR < 25 ml/min/1,73 m2 mag vanwege de beperkte klinische gegevens een behandeling met finerenon niet worden gestart (zie rubriek 4.4 en 5.2).
Voortzetting van de behandeling
Bij patiënten met een eGFR ≥ 15 ml/min/1,73 m2 kan de behandeling met finerenon worden voortgezet met een aanpassing van de dosis op basis van het serumkalium. De eGFR moet 4 weken na het instellen van de behandeling worden gemeten om te bepalen of de startdosis kan worden verhoogd tot de aanbevolen dagelijkse dosis van 20 mg (zie ‘Dosering, Voortzetting van de behandeling’ en tabel 2).
Vanwege beperkte klinische gegevens moet de behandeling met finerenon worden stopgezet bij patiënten die naar het eindstadium nierschade (eGFR < 15 ml/min/1,73 m2) zijn gevorderd (zie rubriek 4.4).
Leverfunctiestoornis
Patiënten met
- ernstige leverfunctiestoornis:
Finerenon mag niet worden gestart (zie rubriek 4.4 en 5.2). Er zijn geen gegevens beschikbaar.
- matige leverfunctiestoornis:
Een initiële dosisaanpassing is niet nodig. Overweeg aanvullende monitoring van het serumkalium en aanpassing van de monitoring aan de hand van de kenmerken van de patiënt (zie rubriek 4.4 en 5.2).
- lichte leverfunctiestoornis:
Een initiële dosisaanpassing is niet nodig.
Gelijktijdige medicatie
Bij patiënten die finerenon gelijktijdig gebruiken met matige of zwakke CYP3A4‑remmers, kaliumsupplementen, trimethoprim of trimethoprim/sulfamethoxazol moeten aanvullende monitoring van het serumkalium en aanpassing van de monitoring aan de hand van de kenmerken van de patiënt worden overwogen (zie rubriek 4.4). Beslissingen over de behandeling met finerenon moeten worden genomen volgens de instructies in tabel 2 (‘Dosering, Voortzetting van de behandeling’).
Het kan noodzakelijk zijn om de behandeling met finerenon tijdelijk te staken, wanneer patiënten trimethoprim of trimethoprim/sulfamethoxazol moeten innemen. Zie rubriek 4.4 en 4.5 voor meer informatie.
Lichaamsgewicht
Een dosisaanpassing op basis van het lichaamsgewicht is niet nodig (zie rubriek 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van finerenon bij kinderen en adolescenten jonger dan 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Oraal gebruik
Tabletten kunnen met een glas water en met of zonder voedsel worden ingenomen (zie rubriek 5.2).
Tabletten mogen niet met grapefruit/pompelmoes of grapefruitsap/pompelmoessap worden ingenomen (zie rubriek 4.5).
Tabletten fijnmaken
Voor patiënten die tabletten niet in hun geheel kunnen doorslikken, mogen Kerendia‑tabletten vlak vóór oraal gebruik worden fijngemaakt en gemengd met water of zachte voeding, zoals appelmoes (zie rubriek 5.2).
4.3 Contra‑indicaties
- Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
- Gelijktijdige behandeling met krachtige CYP3A4‑remmers (zie rubriek 4.5), bijvoorbeeld
- itraconazol
- ketoconazol
- ritonavir
- nelfinavir
- cobicistat
- claritromycine
- telitromycine
- nefazodon
- Ziekte van Addison
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest gemelde bijwerking tijdens behandeling met finerenon was hyperkaliëmie (14,0%). Zie ‘Beschrijving van geselecteerde bijwerkingen, Hyperkaliëmie’ hieronder en rubriek 4.4.
Lijst met bijwerkingen in tabelvorm
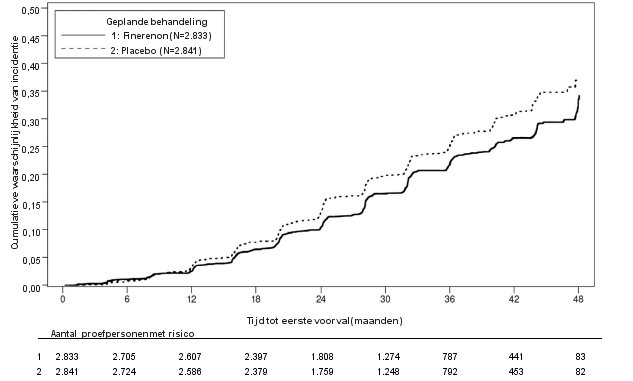
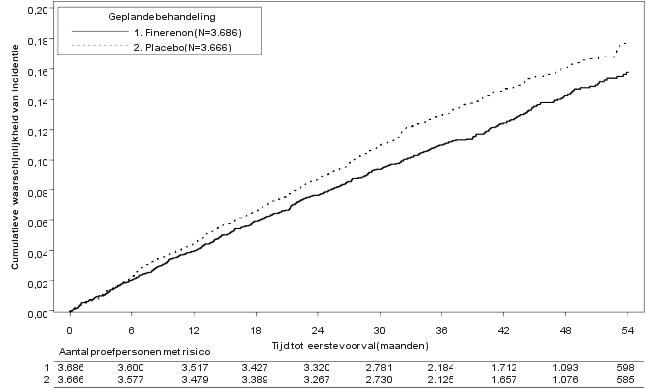
De veiligheid van finerenon bij patiënten met chronische nierschade (chronic kidney disease, CKD) en diabetes mellitus type 2 (DM2) werd geëvalueerd in 2 fase III‑hoofdonderzoeken, FIDELIO‑DKD (diabetische nierschade) en FIGARO‑DKD. In het FIDELIO‑DKD‑onderzoek kregen 2.827 patiënten finerenon (10 of 20 mg eenmaal daags) met een gemiddelde behandelingsduur van 2,2 jaar. In het FIGARO‑DKD‑onderzoek kregen 3.683 patiënten finerenon (10 of 20 mg eenmaal daags) met een gemiddelde behandelingsduur van 2,9 jaar.
De bijwerkingen die werden waargenomen, worden vermeld in tabel 3. Ze worden ingedeeld op basis van de indeling voor systeem/orgaanklassen volgens gegevensbank MedDRA en frequentie.
Bijwerkingen zijn volgens hun frequentie gegroepeerd volgens afnemende ernst.
Frequenties worden als volgt gedefinieerd:
Zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000), niet bekend (kan met de beschikbare gegevens niet worden bepaald).
Tabel 3: Bijwerkingen
Systeem/orgaanklasse | Zeer vaak | Vaak | Soms |
Voedings‑ en stofwisselingsstoornissen | Hyperkaliëmie | Hyponatriëmie |
|
Bloedvataandoeningen |
| Hypotensie |
|
Huid- en onderhuidaandoeningen |
| Pruritus |
|
Onderzoeken |
| Glomerulaire filtratiesnelheid verlaagd | Hemoglobine verlaagd |
Beschrijving van geselecteerde bijwerkingen
Hyperkaliëmie
In de gepoolde gegevens van de FIDELIO‑DKD en FIGARO-DKD‑onderzoeken zijn gevallen van hyperkaliëmie gemeld bij 14,0% van de met finerenon behandelde patiënten in vergelijking met 6,9% van de met placebo behandelde patiënten. In de eerste maand van de behandeling werd in de finerenon‑groep in vergelijking met placebo een stijging van de gemiddelde serumkaliumwaarde met 0,17 mmol/l waargenomen ten opzichte van baseline. De gemiddelde serumkaliumwaarde bleef daarna stabiel. De meeste gevallen van hyperkaliëmie waren licht tot matig van aard en verdwenen bij de met finerenon behandelde patiënten.
Ernstige voorvallen van hyperkaliëmie werden vaker gemeld voor finerenon (1,1%) dan voor placebo (0,2%). Serumkaliumconcentraties van > 5,5 mmol/l en > 6,0 mmol/l zijn gemeld bij respectievelijk 16,8% en 3,3% van de met finerenon behandelde patiënten en bij respectievelijk 7,4% en 1,2% van de met placebo behandelde patiënten. Hyperkaliëmie die leidde tot definitieve stopzetting van de behandeling bij patiënten die finerenon kregen, bedroeg 1,7% tegenover 0,6% bij de placebogroep. Ziekenhuisopname als gevolg van hyperkaliëmie in de finerenon‑groep bedroeg 0,9% tegenover 0,2% in de placebogroep.
Voor specifieke aanbevelingen, zie rubriek 4.2 en 4.4.
Hypotensie
In de gepoolde gegevens van de FIDELIO‑DKD en FIGARO-DKD‑onderzoeken zijn gevallen van hypotensie gemeld bij 4,6% van de met finerenon behandelde patiënten in vergelijking met 3,0% van de met placebo behandelde patiënten. Bij 3 patiënten (< 0,1%) werd de behandeling met finerenon definitief stopgezet vanwege hypotensie. Het aantal gevallen van ziekenhuisopname als gevolg van hypotensie was gelijk voor patiënten die finerenon of placebo kregen (< 0,1%).
De meeste gevallen van hypotensie waren licht of matig van aard en verdwenen bij de met finerenon behandelde patiënten. De gemiddelde systolische bloeddruk daalde in maand 1 met 2‑4 mmHg en de gemiddelde diastolische bloeddruk met 1‑2 mmHg, waarna deze stabiel bleven.
Hyperurikemie
In de gepoolde gegevens van de FIDELIO‑DKD en FIGARO-DKD‑onderzoeken werd hyperurikemie gemeld bij 5,1% van de met finerenon behandelde patiënten in vergelijking met 3,9% van de met placebo behandelde patiënten. Alle voorvallen waren niet‑ernstig en leidden niet tot definitieve stopzetting van de behandeling bij patiënten die finerenon kregen. Vergeleken met placebo werd tot maand 16 in de finerenon‑groep een stijging van de gemiddelde serumurinezuurwaarde van 0,3 mg/dl waargenomen ten opzichte van baseline, die na verloop van tijd afzwakte. Voor de gemelde voorvallen van jicht werd geen verschil waargenomen tussen de finerenon‑groep en de placebogroep (3,0%).
Glomerulaire filtratiesnelheid (GFR) verlaagd
In de gepoolde gegevens van de FIDELIO‑DKD en FIGARO-DKD‑onderzoeken zijn gevallen van verlaagde GFR gemeld bij 5,3% van de met finerenon behandelde patiënten in vergelijking met 4,2% van de met placebo behandelde patiënten. Het aantal gevallen van verlaagde GFR die leidden tot definitieve stopzetting van de behandeling was gelijk voor patiënten die finerenone of placebo kregen (0,2%). Het aantal gevallen van ziekenhuisopname als gevolg van verlaagde GFR was gelijk voor patienten die finerenone of placebo kregen (< 0,1%). De meeste gevallen van verlaagde GFR waren licht of matig van aard en verdwenen bij patiënten die werden behandeld met finerenon. In vergelijking met placebo was bij patiënten die behandeld werden met finerenon sprake van een initiële daling van de eGFR (gemiddeld 2 ml/min/1,73 m2) die na verloop van tijd afzwakte. Deze daling bleek bij ononderbroken voortzetting van de behandeling omkeerbaar te zijn.
Hemoglobine verlaagd
In de gepoolde gegevens van de FIDELIO‑DKD en FIGARO-DKD‑onderzoeken werd finerenon geassocieerd met een door placebo gecorrigeerde absolute afname van gemiddeld hemoglobine van 0,15 g/dl en van gemiddelde hematocriet van 0,5% na 4 maanden van behandeling. Het aantal gerapporteerde gevallen van anemie was vergelijkbaar voor met finerenon behandelde patiënten (6,5%) en de patiënten behandeld met placebo (6,1%). De frequentie van ernstige voorvallen van anemie was laag bij zowel de patiënten behandeld met finerenon en patiënten behandeld met placebo (0,5% . Veranderingen in hemoglobine en hematocriet waren voorbijgaand van aard en bereikten vergelijkbare niveaus met die waargenomen in de met placebo behandelde groep na ongeveer 24‑32 maanden.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via
België
Federaal agentschap voor geneesmiddelen en gezondheidsproducten
Afdeling Vigilantie
Postbus 97
1000 Brussel Madou
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Bayer AG
51368 Leverkusen
Duitsland
8. NUMMERS VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Kerendia 10 mg filmomhulde tabletten
EU/1/21/1616/001-005
Kerendia 20 mg filmomhulde tabletten
EU/1/21/1616/006-010
10. DATUM VAN HERZIENING VAN DE TEKST
02/2023
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4512018 | KERENDIA 10MG FILMOMH TABL 28 | € 72,14 | - | Ja | € 12,8 | € 8,5 | |
| 4512109 | KERENDIA 10MG FILMOMH TABL 98 | € 225,81 | - | Ja | € 15,9 | € 10,5 | |
| 4512117 | KERENDIA 20MG FILMOMH TABL 98 | € 225,81 | - | Ja | € 15,9 | € 10,5 | |
| 4512125 | KERENDIA 20MG FILMOMH TABL 28 | € 72,14 | - | Ja | € 12,8 | € 8,5 |