1. NAAM VAN HET GENEESMIDDEL
Imraldi 40 mg oplossing voor injectie in een voorgevulde spuit.
Imraldi 40 mg oplossing voor injectie in een voorgevulde pen.
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Imraldi 40 mg oplossing voor injectie in een voorgevulde spuit.
Elke voorgevulde spuit van 0,4 ml bevat een enkele dosis van 40 mg adalimumab.
Imraldi 40 mg oplossing voor injectie in een voorgevulde pen.
Elke voorgevulde pen van 0,4 ml bevat een enkele dosis van 40 mg adalimumab.
Adalimumab is een recombinant humaan monoklonaal antilichaam dat wordt geproduceerd in Chinese Hamster Ovariumcellen.
Hulpstof(fen) met bekend effect
Dit geneesmiddel bevat geen hulpstof(fen) met bekend effect.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie (injectievloeistof)
Heldere tot opalescente, kleurloze tot lichtbruine oplossing.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Reumatoïde artritis
Imraldi is in combinatie met methotrexaat geïndiceerd voor:
- de behandeling van volwassen patiënten met matig ernstige tot ernstige, actieve reumatoïde artritis wanneer de respons op langzaam werkende (ziektemodificerende) antireumatische geneesmiddelen, waaronder methotrexaat, ontoereikend is gebleken;
- de behandeling van volwassen patiënten met ernstige, actieve en progressieve reumatoïde artritis die niet eerder behandeld zijn met methotrexaat.
Imraldi kan gegeven worden als monotherapie in geval van intolerantie voor methotrexaat of wanneer voortgezette behandeling met methotrexaat ongewenst is.
Het is aangetoond dat adalimumab de progressie van gewrichtsschade remt, wat gemeten is door middel van röntgenonderzoek, en de fysieke functie verbetert wanneer het gegeven wordt in combinatie met methotrexaat.
Juveniele idiopathische artritis
Polyarticulaire juveniele idiopathische artritis
Imraldi is in combinatie met methotrexaat geïndiceerd voor de behandeling van actieve polyarticulaire juveniele idiopathische artritis bij patiënten vanaf de leeftijd van 2 jaar die een ontoereikende respons hebben gehad op een of meerdere langzaam werkende antireumatische geneesmiddelen (disease-modifying antirheumatic drugs - DMARD’s). Imraldi kan gegeven worden als monotherapie in geval van intolerantie voor methotrexaat of wanneer voortgezette behandeling met methotrexaat ongewenst is (voor de werkzaamheid van monotherapie zie rubriek 5.1). Het gebruik van adalimumab is niet onderzocht bij patiënten jonger dan 2 jaar.
Enthesitis-gerelateerde artritis
Imraldi is geïndiceerd voor de behandeling van actieve enthesitis-gerelateerde artritis bij patiënten vanaf 6 jaar die een ontoereikende respons hebben gehad op conventionele therapie of die conventionele therapie niet verdragen (zie rubriek 5.1).
Axiale spondylartritis
Spondylitis ankylopoetica (AS)
Imraldi is geïndiceerd voor de behandeling van ernstige actieve spondylitis ankylopoetica bij volwassenen die een ontoereikende respons hebben gehad op conventionele therapie.
Axiale spondylartritis zonder röntgenologisch bewijs van AS
Imraldi is geïndiceerd voor de behandeling van volwassenen met ernstige axiale spondylartritis zonder röntgenologisch bewijs van AS, maar met objectieve tekenen van ontsteking door verhoogde CRP en/of MRI, die een ontoereikende respons hebben gehad op of die intolerant zijn voor niet-steroïdale ontstekingsremmers (NSAID’s).
Arthritis psoriatica
Imraldi is geïndiceerd voor de behandeling van actieve en progressieve arthritis psoriatica bij volwassen patiënten wanneer de respons op eerdere therapie met langzaam werkende antireumatische geneesmiddelen ontoereikend is gebleken.
Het is aangetoond dat adalimumab de mate van progressie van perifere gewrichtsschade remt zoals gemeten door middel van röntgenonderzoek bij patiënten met het polyarticulaire symmetrische subtype van de aandoening (zie rubriek 5.1) en dat adalimumab het lichamelijk functioneren verbetert.
Psoriasis
Imraldi is geïndiceerd voor de behandeling van matig ernstige tot ernstige chronische plaquepsoriasis bij volwassen patiënten die in aanmerking komen voor systemische therapie.
Juveniele plaquepsoriasis
Imraldi is geïndiceerd voor de behandeling van ernstige chronische plaquepsoriasis bij kinderen en adolescenten vanaf 4 jaar die een ontoereikende respons hebben gehad op of niet in aanmerking komen voor topicale therapie en lichttherapieën.
Hidradenitis suppurativa (HS)
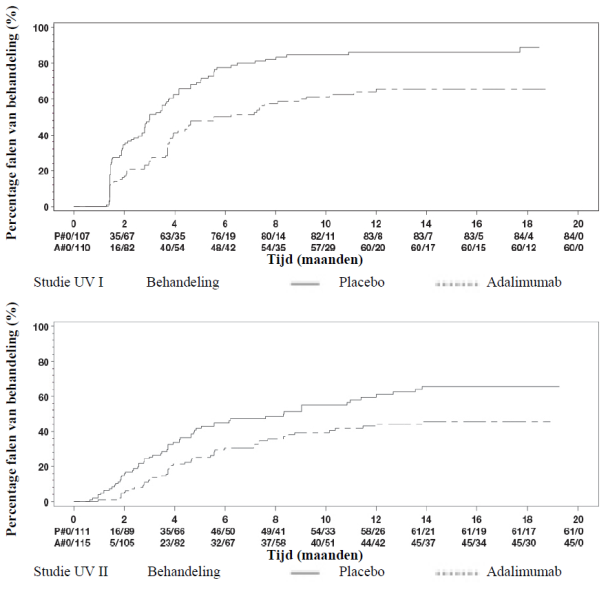
Imraldi is geïndiceerd voor de behandeling van actieve matig ernstige tot ernstige hidradenitis suppurativa (acne inversa) bij volwassenen en adolescenten vanaf 12 jaar met een ontoereikende respons op een conventionele systemische HS-behandeling (zie rubriek 5.1 en 5.2).
De ziekte van Crohn
Imraldi is geïndiceerd voor de behandeling van matig ernstige tot ernstige actieve ziekte van Crohn bij volwassen patiënten die niet gereageerd hebben op een volledige en adequate behandeling met een corticosteroïd en/of een immunosuppressivum, of die dergelijke behandelingen niet verdragen of bij wie hiertegen een contra-indicatie bestaat.
Juveniele ziekte van Crohn
Imraldi is geïndiceerd voor de behandeling van matig ernstige tot ernstige actieve ziekte van Crohn bij kinderen (vanaf 6 jaar) die een ontoereikende respons hebben gehad op conventionele therapie waaronder primaire voedingstherapie en een corticosteroïd en/of een immunomodulator, of die dergelijke behandelingen niet verdragen of bij wie hiertegen een contra-indicatie bestaat.
Colitis ulcerosa
Imraldi is geïndiceerd voor de behandeling van matig ernstige tot ernstige actieve colitis ulcerosa bij volwassen patiënten die een ontoereikende respons hebben gehad op conventionele therapie, waaronder corticosteroïden en 6-mercaptopurine (6-MP) of azathioprine (AZA), of die dergelijke behandelingen niet verdragen of bij wie hiertegen een contra-indicatie bestaat.
Juveniele colitis ulcerosa
Imraldi is geïndiceerd voor de behandeling van matig ernstige tot ernstige actieve colitis ulcerosa bij kinderen (vanaf 6 jaar) die een ontoereikende respons hebben gehad op conventionele behandeling waaronder corticosteroïden en/of 6-mercaptopurine (6-MP) of azathioprine (AZA), of die dergelijke behandelingen niet verdragen of bij wie hiertegen een contra-indicatie bestaat.
Uveïtis
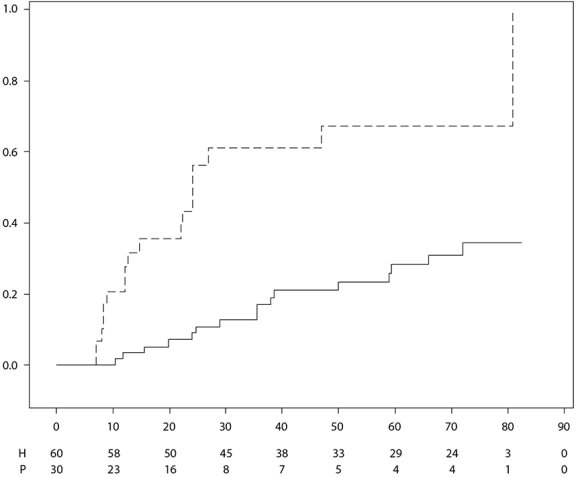
Imraldi is geïndiceerd voor de behandeling van niet-infectieuze intermediaire uveïtis, posterieure uveïtis en panuveïtis bij volwassen patiënten die een ontoereikende respons hebben gehad op corticosteroïden, bij patiënten die minder corticosteroïden moeten gebruiken, of voor wie een corticosteroïde behandeling niet geschikt is.
Juveniele uveïtis
Imraldi is geïndiceerd voor de behandeling van juveniele chronische niet-infectieuze uveïtis anterior bij patiënten vanaf twee jaar die een ontoereikende respons hebben gehad op conventionele behandeling of deze niet verdragen, of voor wie conventionele behandeling niet geschikt is.
4.2 Dosering en wijze van toediening
De Imraldi-behandeling dient te worden geïnitieerd en plaats te vinden onder toezicht van medische specialisten met ervaring in het diagnosticeren en behandelen van de aandoeningen waarvoor Imraldi is geïndiceerd. Oogartsen wordt geadviseerd om te overleggen met een geschikte specialist vóór aanvang van de behandeling met Imraldi (zie rubriek 4.4). Patiënten die behandeld worden met Imraldi moeten de Veiligheidsinformatiekaart voor Patiënten (patiëntenkaart) krijgen.
Na de injectietechniek goed te hebben geoefend, kunnen patiënten zelf Imraldi injecteren als hun arts beslist dat dit passend is, en met medische follow-up voor zover dit nodig is.
Gedurende de behandeling met Imraldi moeten andere gelijktijdige behandelingen (bijv. corticosteroïden en/of immunomodulatoire middelen) worden geoptimaliseerd.
Dosering
Reumatoïde artritis
De aanbevolen dosis Imraldi voor volwassen patiënten met reumatoïde artritis is 40 mg adalimumab eenmaal per twee weken toegediend als een enkele dosis via subcutane injectie. Methotrexaat dient te worden voortgezet tijdens de behandeling met Imraldi.
Glucocorticoïden, salicylaten, niet-steroïdale ontstekingsremmers (NSAID’s) of analgetica kunnen gedurende de behandeling met Imraldi worden gecontinueerd. Aangaande de combinatie met andere langzaam werkende antireumatische geneesmiddelen anders dan methotrexaat zie rubriek 4.4 en 5.1.
Bij gebruik als monotherapie, kunnen patiënten die een afname in hun responshebben op Imraldi 40 mg eenmaal per twee weken baat hebben bij een verhoging van de dosis adalimumab tot 40 mg per week of 80 mg eenmaal per twee weken.
Beschikbare data geven aan dat de klinische respons normaal binnen 12 weken behandeling wordt bereikt. Het vervolgen van de therapie bij patiënten die in deze periode nog niet reageren op het geneesmiddel, dient heroverwogen te worden.
Onderbreking van de toediening
Het kan nodig zijn de toediening te onderbreken, bijvoorbeeld voor een operatie of wanneer een ernstige infectie optreedt.
Beschikbare gegevens suggereren dat het opnieuw starten met adalimumab na stopzetting voor 70 dagen of langer, resulteerde in een even grote klinische respons en een vergelijkbaar veiligheidsprofiel als voor de dosisonderbreking.
Spondylitis ankylopoetica, axiale spondylartritis zonder röntgenologisch bewijs van AS en arthritis psoriatica
De aanbevolen dosis Imraldi voor patiënten met spondylitis ankylopoetica, axiale spondylartritis zonder röntgenologisch bewijs van AS en voor patiënten met arthritis psoriatica is 40 mg adalimumab eenmaal per twee weken toegediend als een enkele dosis via subcutane injectie.
Beschikbare data geven aan dat de klinische respons normaal binnen 12 weken behandeling wordt bereikt. Het vervolgen van de therapie bij patiënten die in deze periode nog niet reageren op het geneesmiddel, dient heroverwogen te worden.
Psoriasis
De aanbevolen dosering Imraldi voor volwassen patiënten bestaat uit een aanvangsdosis van 80 mg, subcutaan toegediend, gevolgd door 40 mg subcutaan eenmaal per twee weken vanaf één week na de aanvangsdosis.
Als een patiënt na 16 weken behandeling nog niet gereageerd heeft, dient voortzetting van de therapie zorgvuldig te worden heroverwogen.
Na 16 weken kunnen patiënten die onvoldoende reageren op Imraldi 40 mg eenmaal per twee weken baat hebben bij een verhoging van de dosis naar 40 mg eenmaal per week of 80 mg eenmaal per twee weken. Bij patiënten met onvoldoende respons op Imraldi dienen de voordelen en risico’s van voortgezette wekelijkse 40 mg behandeling dan wel 80 mg eenmaal per twee weken zorgvuldig te worden afgewogen nadat de dosis is verhoogd (zie rubriek 5.1). Als de respons voldoende is bij 40 mg eenmaal per week of 80 mg eenmaal per twee weken, kan de dosis vervolgens weer naar 40 mg eenmaal per twee weken verlaagd worden.
Hidradenitis suppurativa
Het aanbevolen Imraldi-doseringsschema voor volwassen patiënten met hidradenitis suppurativa (HS) start met 160 mg op dag 1 (kan worden toegediend als vier injecties van 40 mg op één dag of als twee injecties van 40 mg per dag op twee achtereenvolgende dagen), gevolgd door 80 mg twee weken later op dag 15 (toegediend als twee injecties van 40 mg op één dag). Twee weken later (dag 29) wordt de therapie voortgezet met een dosis van 40 mg per week of 80 mg eenmaal per twee weken (toegediend als twee injecties van 40 mg op één dag). Behandelingen met antibiotica mogen indien nodig tijdens de behandeling met Imraldi worden voortgezet. Patiënten wordt aangeraden tijdens de behandeling met Imraldi dagelijks een lokaal antiseptisch middel voor hun HS-laesies te gebruiken.
Als een patiënt na 12 weken behandeling nog niet gereageerd heeft, dient voortzetting van de behandeling zorgvuldig te worden heroverwogen.
Als de behandeling moet worden onderbroken, kan er opnieuw worden gestart met 40 mg Imraldi per week of 80 mg eenmaal per twee weken (zie rubriek 5.1).
De voordelen en risico’s van een aanhoudende langetermijnbehandeling moeten regelmatig geëvalueerd worden (zie rubriek 5.1).
De ziekte van Crohn
Het aanbevolen Imraldi-inductiedoseringsschema voor volwassen patiënten met matig ernstige tot ernstige actieve ziekte van Crohn is 80 mg in week 0, gevolgd door 40 mg in week 2. Indien er een snellere respons op de therapie nodig is, kan het schema 160 mg in week 0 (toegediend als vier 40 mg injecties op één dag of als twee 40 mg injecties per dag voor twee opeenvolgende dagen), gevolgd door 80 mg in week 2 (toegediend als twee 40 mg injecties op één dag) worden gebruikt, waarbij men zich ervan bewust moet zijn dat het risico op bijwerkingen hoger is gedurende de inductie.
Na de inductiebehandeling is de aanbevolen dosering 40 mg eenmaal per twee weken via subcutane injectie. Eventueel mag, indien een patiënt gestopt is met Imraldi en symptomen van de ziekte terugkeren, Imraldi opnieuw worden toegediend. Er is weinig ervaring met opnieuw toedienen na meer dan 8 weken sinds de vorige dosis.
Gedurende de onderhoudsbehandeling, kunnen corticosteroïden geleidelijk worden afgebouwd, overeenkomstig klinische richtlijnen.
Sommige patiënten die een verminderde respons ervaren op Imraldi 40 mg eenmaal per twee weken, kunnen baat hebben bij een verhoging van de dosis naar elke week Imraldi 40 mg of 80 mg eenmaal per twee weken.
Sommige patiënten die geen respons hebben in week 4, kunnen baat hebben bij voortgezette onderhoudsbehandeling tot en met week 12. Voortgezette behandeling dient zorgvuldig te worden heroverwogen bij een patiënt die geen respons ervaart binnen deze periode.
Colitis ulcerosa
Het aanbevolen Imraldi-inductiedoseringsschema voor volwassen patiënten met matig ernstige tot ernstige colitis ulcerosa is 160 mg in week 0 (toegediend als vier 40 mg injecties op één dag of als twee 40 mg injecties per dag voor twee opeenvolgende dagen) en 80 mg in week 2 (toegediend als twee 40 mg injecties op één dag). Na de inductiebehandeling is de aanbevolen dosering 40 mg eenmaal per twee weken via subcutane injectie.
Gedurende de onderhoudsbehandeling kunnen corticosteroïden geleidelijk worden afgebouwd, overeenkomstig klinische richtlijnen.
Sommige patiënten die een verminderde respons ervaren op Imraldi 40 mg eenmaal per twee weken, kunnen baat hebben bij een verhoging van de dosis naar elke week 40 mg Imraldi of 80 mg eenmaal per twee weken.
Beschikbare gegevens tonen aan dat een klinische respons gewoonlijk binnen 2-8 weken behandeling is bereikt. Behandeling met Imraldi dient niet te worden voortgezet bij patiënten die binnen deze periode geen respons ervaren.
Uveïtis
De aanbevolen dosering Imraldi voor volwassen patiënten met uveïtis bestaat uit een aanvangsdosis van 80 mg, gevolgd door 40 mg eenmaal per twee weken vanaf één week na de aanvangsdosis. Er is beperkte ervaring met de start van behandeling met uitsluitend Imraldi. Behandeling met Imraldi kan gestart worden in combinatie met corticosteroïden en/of andere niet-biologische immunomodulatoire middelen. Corticosteroïden die gelijktijdig worden gebruikt, kunnen worden afgebouwd overeenkomstig de klinische praktijk, te beginnen twee weken na aanvang van de behandeling met Imraldi.
Er wordt geadviseerd om de voordelen en risico’s van voortgezette langetermijnbehandeling jaarlijks te evalueren (zie rubriek 5.1).
Speciale populaties
Ouderen
Aanpassing van de dosis is niet vereist.
Nier- en/of leverfunctiestoornis
Adalimumab is niet onderzocht in deze patiëntenpopulaties. Er kan geen doseringsadvies worden gegeven.
Pediatrische patiënten
Imraldi voorgevulde spuit en voorgevulde pen zijn alleen beschikbaar in een dosis van 40 mg. Het is dus niet mogelijk om Imraldi voorgevulde spuit en voorgevulde pen toe te dienen aan pediatrische patiënten die minder dan een volledige dosis van 40 mg nodig hebben. Als er een alternatieve dosis nodig is, moeten andere aanbiedingsvormen worden gebruikt die een dergelijke optie bieden.
Juveniele idiopathische artritis
Polyarticulaire juveniele idiopathische artritis vanaf de leeftijd van 2 jaar
De aanbevolen dosis Imraldi voor patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd vanaf 2 jaar is gebaseerd op het lichaamsgewicht (tabel 1). Imraldi wordt eenmaal per twee weken toegediend via subcutane injectie.
Tabel 1. Imraldi dosis voor patiënten met polyarticulaire juveniele idiopathische artritis
Patiëntgewicht | Doseringsschema |
10 kg tot < 30 kg | 20 mg eenmaal per twee weken |
≥ 30 kg | 40 mg eenmaal per twee weken |
Beschikbare gegevens duiden erop dat de klinische respons meestal binnen 12 weken behandeling wordt bereikt. Voortgezette behandeling dient zorgvuldig te worden heroverwogen bij een patiënt die geen respons ervaart binnen deze periode.
Er is geen relevante toepassing van adalimumab bij patiënten jonger dan 2 jaar voor deze indicatie.
Enthesitis-gerelateerde artritis
De aanbevolen dosis Imraldi voor patiënten met enthesitis-gerelateerde artritis met een leeftijd van 6 jaar en ouder is gebaseerd op het lichaamsgewicht (tabel 2). Imraldi wordt eenmaal per twee weken toegediend via subcutane injectie.
Tabel 2. Imraldi dosis voor patiënten met enthesitis-gerelateerde artritis
Patiëntgewicht | Doseringsschema |
15 kg tot < 30 kg | 20 mg eenmaal per twee weken |
≥ 30 kg | 40 mg eenmaal per twee weken |
Het gebruik van adalimumab is niet onderzocht bij patiënten met enthesitis-gerelateerde artritis jonger dan 6 jaar.
Juveniele plaquepsoriasis
De aanbevolen dosis Imraldi voor patiënten met plaquepsoriasis van 4 tot en met 17 jaar oud is gebaseerd op lichaamsgewicht (tabel 3). Imraldi wordt toegediend via subcutane injectie.
Tabel 3. Imraldi dosis voor kinderen met plaquepsoriasis
Patiëntgewicht | Doseringsschema |
15 kg tot < 30 kg | Aanvangsdosis van 20 mg, gevolgd door 20 mg eenmaal per twee weken vanaf één week na de aanvangsdosis |
≥ 30 kg | Aanvangsdosis van 40 mg, gevolgd door 40 mg eenmaal per twee weken vanaf één week na de aanvangsdosis |
Voortgezette behandeling dient zorgvuldig te worden heroverwogen bij een patiënt die geen respons ervaart binnen 16 weken.
Als herbehandeling met Imraldi geïndiceerd is, dient bovenstaande aanbeveling over de dosering en de behandelingsduur gevolgd te worden.
De veiligheid van adalimumab bij kinderen met plaquepsoriasis is beoordeeld gedurende gemiddeld 13 maanden.
Er is geen relevante toepassing van adalimumab bij kinderen jonger dan 4 jaar voor deze indicatie.
Hidradenitis suppurativa bij adolescenten (vanaf de leeftijd van 12 jaar met een gewicht van minstens 30 kg)
Er zijn geen klinische onderzoeken met adalimumab bij adolescente patiënten met HS. De dosering van adalimumab bij deze patiënten is vastgesteld op basis van farmacokinetische modellering en simulatie (zie rubriek 5.2).
De aanbevolen dosis Imraldi is 80 mg in week 0, gevolgd door 40 mg eenmaal per twee weken, waarbij in week 1 wordt gestart met subcutane injectie.
Bij adolescente patiënten met een ontoereikende respons op Imraldi 40 mg eenmaal per twee weken kan een verhoging van de dosis naar 40 mg per week of 80 mg eenmaal per twee weken worden overwogen.
Het gebruik van antibiotica mag tijdens de behandeling met Imraldi indien nodig worden voortgezet. Patiënten wordt aangeraden tijdens de behandeling met Imraldi dagelijks een lokaal antiseptisch middel voor hun HS-laesies te gebruiken.
Voortzetting van de behandeling na 12 weken dient zorgvuldig heroverwogen te worden bij een patiënt die binnen deze periode geen verbetering laat zien.
Als de behandeling moet worden onderbroken, kan er indien relevant opnieuw met Imraldi worden gestart.
De voordelen en risico's van een aanhoudende langetermijnbehandeling moeten regelmatig geëvalueerd worden (zie gegevens over volwassenen in rubriek 5.1).
Er is geen relevante toepassing van adalimumab bij kinderen jonger dan 12 jaar bij deze indicatie.
Juveniele ziekte van Crohn
De aanbevolen dosis Imraldi voor patiënten met de ziekte van Crohn van 6 tot en met 17 jaar oud is gebaseerd op lichaamsgewicht (tabel 4). Imraldi wordt toegediend via subcutane injectie.
Tabel 4. Imraldi dosis voor kinderen met de ziekte van Crohn
Patiënt-gewicht | Inductiedosering | Onderhoudsdosering |
< 40 kg |
| 20 mg eenmaal per twee weken |
≥ 40 kg |
| 40 mg eenmaal per twee weken |
Patiënten die onvoldoende respons ervaren, kunnen baat hebben bij een verhoging van de dosis:
- < 40 kg: 20 mg eenmaal per week
- ≥ 40 kg: 40 mg eenmaal per week of 80 mg eenmaal per twee weken
Voortzetting van de behandeling dient zorgvuldig te worden overwogen wanneer een patiënt in week 12 nog geen respons vertoont.
Er is geen relevante toepassing van adalimumab bij kinderen jonger dan 6 jaar voor deze indicatie.
Juveniele colitis ulcerosa
De aanbevolen dosis Imraldi voor patiënten van 6 tot en met 17 jaar met colitis ulcerosa is gebaseerd op het lichaamsgewicht (tabel 5). Imraldi wordt toegediend via subcutane injectie.
Tabel 5. Imraldi dosis voor pediatrische patiënten met colitis ulcerosa
Patiëntgewicht | Inductiedosering | Onderhoudsdosering |
< 40 kg |
|
|
≥ 40 kg |
|
|
* Pediatrische patiënten die 18 jaar worden tijdens behandeling met Imraldi dienen door te gaan met de hun voorgeschreven onderhoudsdosis.
Voortzetting van de behandeling na 8 weken dient zorgvuldig te worden overwogen bij patiënten die geen tekenen van een respons vertonen binnen deze tijdsperiode.
Er is geen relevante toepassing van Imraldi bij kinderen jonger dan 6 jaar voor deze indicatie.
Imraldi kan in andere sterkten en/of toedieningsvormen beschikbaar zijn, afhankelijk van de individuele behandelingsbehoeften.
Arthritis psoriatica en axiale spondylartritis waaronder spondylitis ankylopoetica
Er is geen relevante toepassing van adalimumab bij pediatrische patiënten voor de indicaties spondylitis ankylopoetica en arthritis psoriatica.
Juveniele uveïtis
De aanbevolen dosis Imraldi voor kinderen met uveïtis vanaf 2 jaar is gebaseerd op het lichaamsgewicht (tabel 6). Imraldi wordt toegediend via subcutane injectie.
Voor juveniele uveïtis is er geen ervaring met de behandeling van adalimumab zonder gelijktijdig gebruik van methotrexaat.
Tabel 6. Imraldi dosis voor kinderen met uveïtis
Patiëntgewicht | Doseringsschema |
< 30 kg | 20 mg eenmaal per twee weken in combinatie met methotrexaat |
≥ 30 kg | 40 mg eenmaal per twee weken in combinatie met methotrexaat |
Bij initiatie van de Imraldi-behandeling kan één week voor aanvang van de onderhoudsbehandeling een oplaaddosis van 40 mg worden toegediend voor patiënten < 30 kg of 80 mg voor patiënten ≥ 30 kg. Er zijn geen klinische gegevens beschikbaar over het gebruik van een oplaaddosis adalimumab bij kinderen jonger dan 6 jaar (zie rubriek 5.2).
Er is geen relevante toepassing van adalimumab bij kinderen jonger dan 2 jaar voor deze indicatie.
De verhouding tussen voordelen en risico’s van voortgezette langetermijnbehandeling moet jaarlijks geëvalueerd worden (zie rubriek 5.1).
Wijze van toediening
Imraldi wordt toegediend via subcutane injectie. Een volledige gebruiksaanwijzing is te vinden in de bijsluiter.
Een voorgevulde spuit en voorgevulde pen van 40 mg zijn beschikbaar voor patiënten voor het toedienen van een volledige dosis van 40 mg.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Actieve tuberculose of andere ernstige infecties, zoals sepsis, en andere opportunistische infecties (zie rubriek 4.4).
Matig ernstig tot ernstig hartfalen (NYHA-klasse III/IV) (zie rubriek 4.4).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Adalimumab is tot 60 maanden of langer onderzocht bij 9.506 patiënten in de belangrijkste gecontroleerde en open-label onderzoeken. Bij deze onderzoeken waren patiënten betrokken met kort bestaande en langer bestaande reumatoïde artritis, met juveniele idiopathische artritis (polyarticulaire juveniele idiopathische artritis en enthesitis-gerelateerde artritis) en met axiale spondylartritis (spondylitis ankylopoetica en axiale spondylartritis zonder röntgenologisch bewijs van AS), arthritis psoriatica, de ziekte van Crohn, colitis ulcerosa, psoriasis, hidradenitis suppurativa en uveïtis. In de belangrijkste gecontroleerde onderzoeken kregen 6.089 patiënten adalimumab en 3.801 patiënten een placebo of actief controlemiddel tijdens de gecontroleerde periode.
Het deel van de patiënten dat de behandeling staakte omwille van bijwerkingen tijdens het dubbelblinde gecontroleerde deel van de belangrijkste onderzoeken bedroeg 5,9% voor de patiënten die adalimumab gebruikten en 5,4% voor met het controlemiddel behandelde patiënten.
De meest gemelde bijwerkingen zijn infecties (zoals nasofaryngitis, infectie van de bovenste luchtwegen en sinusitis), reacties op de injectieplaats (erytheem, jeuk, bloeding, pijn of zwelling), hoofdpijn en pijn van het skeletspierstelsel.
Voor adalimumab zijn meldingen van ernstige bijwerkingen gedaan. TNF-antagonisten zoals adalimumab hebben een effect op het immuunsysteem en het gebruik ervan kan de afweer van het lichaam tegen infecties en kanker beïnvloeden.
Fatale en levensbedreigende infecties (waaronder sepsis, opportunistische infecties en TB), HBV-reactivatie en verscheidene maligniteiten (waaronder leukemie, lymfomen en HSTCL) zijn ook gemeld bij gebruik van adalimumab.
Ook zijn meldingen gedaan van ernstige hematologische, neurologische en auto-immuunreacties. Deze omvatten zeldzame gevallen van pancytopenie, aplastische anemie, centrale en perifere demyeliniserende aandoeningen en meldingen van lupus, lupus-gerelateerde aandoeningen en Stevens-Johnson-syndroom.
Pediatrische patiënten
In het algemeen waren de bijwerkingen bij kinderen qua frequentie en type vergelijkbaar met de bij volwassen patiënten waargenomen bijwerkingen.
Getabelleerde lijst van bijwerkingen
De vermelde lijst met bijwerkingen is gebaseerd op ervaring uit klinische studies en op postmarketingervaring en is weergegeven per systeem/orgaanklasse en frequentie hieronder in tabel 7: zeer vaak ( 1/10); vaak ( 1/100, < 1/10); soms (≥ 1/1.000, < 1/100); zelden (≥ 1/10.000, < 1/1.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen iedere frequentiegroep worden bijwerkingen gerangschikt naar afnemende ernst. De hoogste frequentie die werd waargenomen bij de verschillende indicaties is opgenomen. Een asterisk (*) in de ‘Systeem/orgaanklasse’-kolom betekent dat aanvullende informatie elders in rubriek 4.3, 4.4 en 4.8 gevonden kan worden.
Tabel 7
Bijwerkingen
Systeem/orgaanklasse | Frequentie | Bijwerking |
Infecties en parasitaire aandoeningen* | zeer vaak | luchtweginfecties (waaronder lagere en hogere luchtweginfecties, pneumonie, sinusitis, faryngitis, nasofaryngitis en virale herpes pneumonie) |
vaak | systemische infecties (waaronder sepsis, candidiasis en influenza), | |
soms | neurologische infecties (waaronder virale meningitis), opportunistische infecties en tuberculose (waaronder coccidioïdomycose, histoplasmose en MAC-infectie (Mycobacterium avium complex)), | |
Neoplasmata, benigne, maligne en niet-gespecificeerd (inclusief cysten en poliepen)* | vaak | huidkanker met uitzondering van melanoom (waaronder basaalcelcarcinoom en squameuzecelcarcinoom), |
soms | lymfoom**, | |
zelden | leukemie1) | |
niet bekend | hepatosplenisch T-cellymfoom1) | |
Bloed- en lymfestelselaandoeningen* | zeer vaak | leukopenie (waaronder neutropenie en agranulocytose), |
vaak | leukocytose, | |
soms | idiopathische trombocytopenische purpura | |
zelden | pancytopenie | |
Immuunsysteemaandoeningen* | vaak | overgevoeligheid, |
soms | sarcoïdose1), | |
zelden | anafylaxie1) | |
Voedings- en stofwisselingsstoornissen | zeer vaak | verhoogde lipiden |
vaak | hypokaliëmie, | |
Psychische stoornissen | vaak | stemmingswisselingen (waaronder depressie), |
Zenuwstelselaandoeningen* | zeer vaak | hoofdpijn |
vaak | paresthesieën (waaronder hypo-esthesie), | |
soms | cerebrovasculair accident1), | |
zelden | multipele sclerose, | |
Oogaandoeningen | vaak | visusstoornis, |
soms | diplopie | |
Evenwichtsorgaan- en ooraandoeningen | vaak | vertigo |
soms | doofheid, | |
Hartaandoeningen* | vaak | tachycardie |
soms | myocardinfarct1), | |
zelden | hartstilstand | |
Bloedvataandoeningen | vaak | hypertensie, |
soms | aneurysma aortae, | |
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen* | vaak | astma, |
soms | longembolie1), | |
zelden | longfibrose1) | |
Maagdarmstelselaandoeningen | zeer vaak | buikpijn, misselijkheid en braken |
vaak | maag-darmbloeding, | |
soms | pancreatitis, | |
zelden | intestinale perforatie1) | |
Lever- en galaandoeningen* | zeer vaak | verhoogde leverenzymen |
soms | cholecystitis en cholelithiase, | |
zelden | hepatitis | |
niet bekend | leverfalen1) | |
Huid- en onderhuidaandoeningen | zeer vaak | rash (waaronder schilferende rash) |
vaak | verergering of het ontstaan van psoriasis (inclusief psoriasis pustulosa palmoplantaris)1), | |
soms | nachtzweten, | |
zelden | erythema multiforme1), | |
niet bekend | verergering van symptomen van dermatomyositis1) | |
Skeletspierstelsel- en bindweefselaandoeningen | zeer vaak | pijn van het skeletspierstelsel |
vaak | spierspasmen (waaronder verhoging van de hoeveelheid creatinefosfokinase in het bloed) | |
soms | rabdomyolyse, | |
zelden | lupus-achtig syndroom1) | |
Nier- en urinewegaandoeningen | vaak | nierfunctiestoornissen, hematurie |
soms | nycturie | |
Voortplantingsstelsel- en borstaandoeningen | soms | erectiele disfunctie |
Algemene aandoeningen en toedieningsplaatsstoornissen* | zeer vaak | reacties op de injectieplaats (waaronder erytheem op de injectieplaats) |
vaak | pijn op de borst, | |
soms | ontsteking | |
Onderzoeken* | vaak | stollings- en bloedingsstoornissen (waaronder verlengde geactiveerde partiële tromboplastinetijd), positieve test op autoantilichamen (waaronder antilichamen tegen dubbelstrengs DNA), |
niet bekend | gewichtstoename2) | |
Letsels, intoxicaties en verrichtingscomplicaties | vaak | gestoorde genezing |
* nadere informatie is elders in rubriek 4.3, 4.4 en 4.8 te vinden
** inclusief aanvullende open-label onderzoeken
1) inclusief spontane meldingen
2) De gemiddelde gewichtsverandering vanaf baseline voor adalimumab varieerde van 0,3 kg tot 1,0 kg voor de verschillende indicaties voor volwassenen ten opzichte van (minus) -0,4 kg tot 0,4 kg voor placebo gedurende een behandelperiode van 4-6 maanden. Er werd ook een gewichtstoename van 5-6 kg waargenomen in langlopende verlengingsonderzoeken met een gemiddelde blootstelling van ongeveer 1-2 jaar zonder controlegroep, met name bij patiënten met ziekte van Crohn en colitis ulcerosa. Het mechanisme achter dit effect is onduidelijk, maar zou verband kunnen houden met het ontstekingsremmende effect van adalimumab
Hidradenitis suppurativa (HS)
Het veiligheidsprofiel voor HS-patiënten die eenmaal per week met adalimumab werden behandeld, kwam overeen met het reeds bekende veiligheidsprofiel van adalimumab.
Uveïtis
Het veiligheidsprofiel voor patiënten met uveïtis die eenmaal per twee weken met adalimumab werden behandeld, kwam overeen met het reeds bekende veiligheidsprofiel van adalimumab.
Beschrijving van geselecteerde bijwerkingen
Reacties op de injectieplaats
In de belangrijkste gecontroleerde onderzoeken bij volwassenen en kinderen traden bij 12,9% van de met adalimumab behandelde patiënten reacties op de injectieplaats op (erytheem en/of jeuk, bloeding, pijn of zwelling), in vergelijking met 7,2% van de patiënten die placebo of actief controlemiddel kregen. Reacties op de injectieplaats noodzaakten doorgaans niet tot staken van het geneesmiddel.
Infecties
In de belangrijkste gecontroleerde onderzoeken bij volwassenen en kinderen bedroeg het incidentiecijfer voor infectie 1,51 per patiëntjaar bij de met adalimumab behandelde patiënten en 1,46 per patiëntjaar bij de met placebo en actief controlemiddel behandelde patiënten. De infecties bestonden hoofdzakelijk uit nasofaryngitis, bovensteluchtweginfecties en sinusitis. De meeste patiënten bleven op adalimumab na het verdwijnen van de infectie.
De incidentie van ernstige infecties bedroeg 0,04 per patiëntjaar bij met adalimumab behandelde patiënten en 0,03 per patiëntjaar bij met placebo en actief controlemiddel behandelde patiënten.
In gecontroleerde en open-label onderzoeken met adalimumab bij volwassenen en kinderen zijn ernstige infecties (waaronder fatale infecties, die zelden voorkwamen) gemeld, waaronder tuberculose (inclusief miliaire en extrapulmonale locaties) en invasieve opportunistische infecties (bijv. gedissemineerde of extrapulmonaire histoplasmose, blastomycose, coccidioïdomycose, pneumocystose, candidiasis, aspergillose en listeriose). De meeste gevallen van tuberculose traden op in de eerste acht maanden na het starten van de therapie en kunnen duiden op een recidief van een latente ziekte.
Maligniteiten en lymfoproliferatieve aandoeningen
Er zijn geen maligniteiten waargenomen bij 249 pediatrische patiënten met een blootstelling van 655,6 patiëntjaren tijdens onderzoeken met adalimumab bij patiënten met juveniele idiopathische artritis (polyarticulaire juveniele idiopathische artritis en enthesitis-gerelateerde artritis). Daarnaast zijn er geen maligniteiten waargenomen bij 192 pediatrische patiënten met een blootstelling van 498,1 patiëntjaren tijdens onderzoeken met adalimumab in kinderen met de ziekte van Crohn. Er zijn geen maligniteiten waargenomen bij 77 pediatrische patiënten met een blootstelling van 80,0 patiëntjaren tijdens een onderzoek met adalimumab bij pediatrische patiënten met chronische plaquepsoriasis. Er zijn geen maligniteiten waargenomen bij 93 pediatrische patiënten met een blootstelling van 65,3 patiëntjaren tijdens een onderzoek met adalimumab bij pediatrische patiënten met colitis ulcerosa. Er zijn geen maligniteiten waargenomen bij 60 pediatrische patiënten met een blootstelling van 58,4 patiëntjaren tijdens een onderzoek met adalimumab bij pediatrische patiënten met uveïtis.
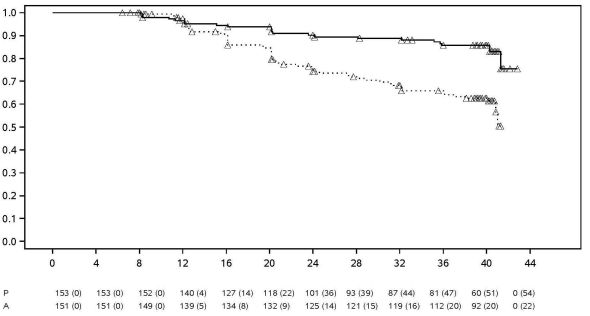
Tijdens de gecontroleerde gedeelten van belangrijke adalimumab-onderzoeken bij volwassenen die ten minste 12 weken duurden bij patiënten met matig ernstige tot ernstige actieve reumatoïde artritis, spondylitis ankylopoetica, axiale spondylartritis zonder röntgenologisch bewijs van AS, arthritis psoriatica, psoriasis, hidradenitis suppurativa, de ziekte van Crohn, colitis ulcerosa en uveïtis werden maligniteiten, anders dan lymfomen en niet-melanoom huidkanker, waargenomen met een incidentie (95% betrouwbaarheidsinterval) van 6,8 (4,4; 10,5) per 1.000 patiëntjaren bij 5.291 met adalimumab behandelde patiënten versus een incidentie van 6,3 (3,4; 11,8) per 1.000 patiëntjaren bij 3.444 controlepatiënten (mediane behandelingsduur was 4,0 maanden voor adalimumab en 3,8 maanden voor de controlepatiënten). De incidentie (95% betrouwbaarheidsinterval) van niet-melanoom huidcarcinomen was 8,8 (6,0; 13,0) per 1.000 patiëntjaren bij de met adalimumab behandelde patiënten en 3,2 (1,3; 7,6) per 1.000 patiëntjaren bij de controlepatiënten. Van deze huidcarcinomen, bedroeg de incidentie (95% betrouwbaarheidsinterval) van plaveiselcelcarcinoom 2,7 (1,4; 5,4) per 1.000 patiëntjaren bij met adalimumab behandelde patiënten en 0,6 (0,1; 4,5) per 1.000 patiëntjaren bij de controlepatiënten. De incidentie (95% betrouwbaarheidsinterval) van lymfomen bedroeg 0,7 (0,2; 2,7) per 1.000 patiëntjaren bij met adalimumab behandelde patiënten en 0,6 (0,1; 4,5) per 1.000 patiëntjaren bij de controlepatiënten.
Bij het combineren van de gecontroleerde gedeelten van deze onderzoeken en de lopende en afgeronde open-label extensieonderzoeken met een mediane duur van ongeveer 3,3 jaar waarin 6.427 patiënten geïncludeerd waren en meer dan 26.439 patiëntjaren van therapie, is het waargenomen aantal maligniteiten, anders dan lymfomen en niet-melanoom huidcarcinomen ongeveer 8,5 per 1.000 patiëntjaren. De waargenomen incidentie van niet-melanoom huidcarcinomen bedraagt ongeveer 9,6 per 1.000 patiëntjaren en voor lymfomen ongeveer 1,3 per 1.000 patiëntjaren.
Tijdens postmarketing ervaringen van januari 2003 tot december 2010, voornamelijk bij patiënten met reumatoïde artritis, was de gemelde incidentie van spontaan gerapporteerde maligniteiten ongeveer 2,7 per 1.000 patiëntbehandeljaren. De spontaan gerapporteerde incidenties van niet-melanoom huidcarcinomen en lymfomen waren respectievelijk ongeveer 0,2 en 0,3 per 1.000 patiëntbehandeljaren (zie rubriek 4.4).
Zeldzame postmarketinggevallen van hepatosplenisch T-cellymfoom zijn gerapporteerd bij patiënten die behandeld werden met adalimumab (zie rubriek 4.4).
Autoantilichamen
Op verschillende tijdstippen tijdens de onderzoeken met reumatoïde artritis I-V werden serummonsters van de patiënten getest op autoantilichamen. In deze onderzoeken werden voor 11,9% van de met adalimumab behandelde patiënten en 8,1% van de met placebo en actief controlemiddel behandelde patiënten die aan het begin van het onderzoek negatieve antinucleaire-antilichaamtiters hadden, positieve titers gemeld in week 24. Twee van de 3.441 met adalimumab behandelde patiënten in alle onderzoeken met reumatoïde artritis en arthritis psoriatica vertoonden klinische symptomen die wezen op recent opgetreden lupusachtig syndroom. De patiënten vertoonden verbetering na het staken van de behandeling. Er waren geen patiënten bij wie lupusnefritis of symptomen van het centrale zenuwstelsel optraden.
Lever- en galaandoeningen
In de gecontroleerde klinische fase 3-onderzoeken met adalimumab bij patiënten met reumatoïde artritis en arthritis psoriatica met een controleperiode met een duur variërend van 4 tot 104 weken, kwamen ALAT-verhogingen van ≥ 3 × ULN voor bij 3,7% van de patiënten die werden behandeld met adalimumab en bij 1,6% van de patiënten in de controle-arm.
In de gecontroleerde klinische fase 3-onderzoeken met adalimumab bij patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd van 4 tot en met 17 jaar en enthesitis-gerelateerde artritis in de leeftijd van 6 tot en met 17 jaar, kwamen ALAT-verhogingen van ≥ 3 × ULN voor bij 6,1% van de patiënten die werden behandeld met adalimumab en bij 1,3% van de patiënten in de controle-arm. De meeste ALAT-verhogingen kwamen voor tijdens gelijktijdig gebruik van methotrexaat. In het klinische fase 3-onderzoek met adalimumab kwamen geen ALAT-verhogingen van ≥ 3 × ULN voor bij patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd van 2 tot 4 jaar.
In de gecontroleerde klinische fase 3-onderzoeken met adalimumab bij patiënten met de ziekte van Crohn en colitis ulcerosa waarbij de controleperiode varieerde van 4 tot 52 weken, kwamen ALAT-verhogingen van ≥ 3 × ULN voor bij 0,9% van de patiënten die werden behandeld met adalimumab en bij 0,9% van de patiënten in de controle-arm.
In het fase 3-onderzoek met adalimumab werden bij patiënten met juveniele ziekte van Crohn de werkzaamheid en veiligheid tot 52 weken behandeling beoordeeld van twee op lichaamsgewicht aangepaste onderhoudsdoseringregimes na een op lichaamsgewicht aangepaste inductietherapie. Hierbij kwamen ALAT-verhogingen van ≥ 3 × ULN voor bij 2,6% (5/192) van de patiënten van wie er 4 in de uitgangssituatie gelijktijdig immunosuppressiva toegediend kregen.
In de gecontroleerde klinische fase 3-onderzoeken met adalimumab bij patiënten met plaquepsoriasis waarbij de controleperiode varieerde van 12 tot 24 weken, kwamen ALAT-verhogingen van ≥ 3 × ULN voor bij 1,8% van de patiënten die werden behandeld met adalimumab en bij 1,8% van de patiënten in de controle-arm.
Er kwamen geen ALAT-verhogingen van ≥ 3 × ULN voor in het fase 3-onderzoek met adalimumab bij pediatrische patiënten met plaquepsoriasis.
In gecontroleerde onderzoeken kregen patiënten met hidradenitis suppurativa adalimumab (toegediend in initiële doses van 160 mg in week 0 en 80 mg in week 2, gevolgd door wekelijkse doses van 40 mg vanaf week 4) waarbij de controleperiode varieerde van 12 tot 16 weken. ALAT-verhogingen van ≥ 3 × ULN kwamen voor bij 0,3% van de patiënten die werden behandeld met adalimumab en bij 0,6% van de patiënten in de controle-arm.
In gecontroleerde onderzoeken kregen volwassen patiënten met uveïtis adalimumab (initiële doses van 80 mg in week 0, gevolgd door 40 mg eenmaal per twee weken vanaf week 1) tot 80 weken met een mediane blootstelling van 166,5 dagen en 105,0 dagen bij respectievelijk patiënten die werden behandeld met adalimumab en patiënten in de controle-arm. ALAT-verhogingen van ≥ 3 × ULN kwamen hierbij voor bij 2,4% van de patiënten die werden behandeld met adalimumab en bij 2,4% van de patiënten in de controle-arm.
In het gecontroleerde fase 3-onderzoek van adalimumab bij patiënten met juveniele colitis ulcerosa (N = 93) waarin de werkzaamheid en veiligheid werden beoordeeld van een onderhoudsdosering van 0,6 mg/kg (maximaal 40 mg) eenmaal per twee weken (N = 31) en een onderhoudsdosering van 0,6 mg/kg (maximaal 40 mg) eenmaal per week (N = 32), volgend op een voor lichaamsgewicht gecorrigeerde inductiedosering van 2,4 mg/kg (maximaal 160 mg) in week 0 en week 1 en 1,2 mg/kg (maximaal 80 mg) in week 2 (N = 63), of een inductiedosering van 2,4 mg/kg (maximaal 160 mg) in week 0, placebo in week 1 en 1,2 mg/kg (maximaal 80 mg) in week 2 (N = 30), kwamen ALAT-verhogingen van ≥ 3 x ULN voor bij 1,1% (1/93) van de patiënten.
Bij de klinische onderzoeken van alle indicaties waren patiënten met een verhoogd ALAT klachtenvrij en in de meeste gevallen waren de verhogingen voorbijgaand van aard en verdwenen gedurende de voortzetting van de behandeling. Er zijn echter ook postmarketingmeldingen van leverfalen, evenals minder ernstige leveraandoeningen die kunnen voorafgaan aan leverfalen, zoals hepatitis waaronder auto-immuunhepatitis bij patiënten die adalimumab kregen.
Gelijktijdige behandeling met azathioprine/6-mercaptopurine
Tijdens onderzoeken bij volwassenen met de ziekte van Crohn werden hogere incidenties van maligne en ernstige infectiegerelateerde bijwerkingen gezien bij de combinatie van adalimumab en azathioprine/6-mercaptopurine in vergelijking met alleen adalimumab.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via
Nederland
Nederlands Bijwerkingen Centrum Lareb
Website: www.lareb.nl
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Samsung Bioepis NL B.V.
Olof Palmestraat 10
2616 LR Delft
Nederland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Imraldi 40 mg oplossing voor injectie in een voorgevulde spuit
EU/1/17/1216/010
EU/1/17/1216/011
EU/1/17/1216/012
EU/1/17/1216/013
Imraldi 40 mg oplossing voor injectie in een voorgevulde pen
EU/1/17/1216/014
EU/1/17/1216/015
EU/1/17/1216/016
EU/1/17/1216/017
10. DATUM VAN HERZIENING VAN DE TEKST
09/2024
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4613360 | IMRALDI 40MG/0,4ML OPL INJ 100MG/ML VOORGEV.SPUIT2 | L04AB04 | € 452,85 | - | Ja | € 12,8 | € 8,5 |
| 4613378 | IMRALDI 40MG/0,4ML OPL INJ 100MG/ML VOORGEV.SPUIT6 | L04AB04 | € 1357,57 | - | Ja | € 12,8 | € 8,5 |
| 4613386 | IMRALDI 40MG/0,4ML OPL INJ 100MG/ML VOORGEV. PEN 2 | L04AB04 | € 452,85 | - | Ja | € 12,8 | € 8,5 |
| 4613394 | IMRALDI 40MG/0,4ML OPL INJ 100MG/ML VOORGEV. PEN 6 | L04AB04 | € 1357,57 | - | Ja | € 12,8 | € 8,5 |