1. NAAM VAN HET GENEESMIDDEL
Mayzent 0,25 mg filmomhulde tabletten
Mayzent 1 mg filmomhulde tabletten
Mayzent 2 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Mayzent 0,25 mg filmomhulde tabletten
Elke filmomhulde tablet bevat siponimod-fumaarzuur overeenkomend met 0,25 mg siponimod.
Hulpstoffen met bekend effect
Elke tablet bevat 59,1 mg lactose (als monohydraat) en 0,092 mg sojalecithine.
Mayzent 1 mg filmomhulde tabletten
Elke filmomhulde tablet bevat siponimod-fumaarzuur overeenkomend met 1 mg siponimod.
Hulpstoffen met bekend effect
Elke tablet bevat 58,3 mg lactose (als monohydraat) en 0,092 mg sojalecithine.
Mayzent 2 mg filmomhulde tabletten
Elke filmomhulde tablet bevat siponimod-fumaarzuur overeenkomend met 2 mg siponimod.
Hulpstoffen met bekend effect
Elke tablet bevat 57,3 mg lactose (als monohydraat) en 0,092 mg sojalecithine.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet
Mayzent 0,25 mg filmomhulde tabletten
Lichtrode, ronde, biconvexe filmomhulde tablet met schuine rand en een diameter van ongeveer 6,1 mm met het bedrijfslogo op de ene zijde en “T” op de andere zijde.
Mayzent 1 mg filmomhulde tabletten
Paarswitte, ronde, biconvexe filmomhulde tablet met schuine rand en een diameter van ongeveer 6,1 mm met het bedrijfslogo op de ene zijde en “L” op de andere zijde.
Mayzent 2 mg filmomhulde tabletten
Lichtgele, ronde, biconvexe filmomhulde tablet met schuine rand en een diameter van ongeveer 6,1 mm met het bedrijfslogo op de ene zijde en “II” op de andere zijde.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
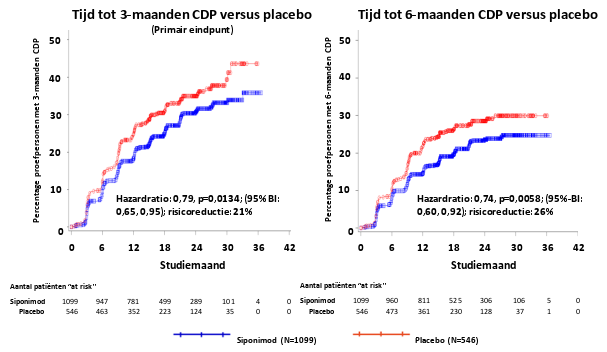
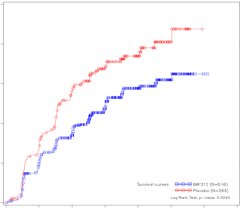
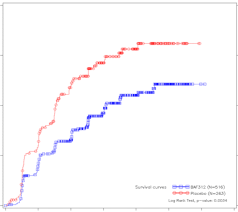
Mayzent is geïndiceerd voor de behandeling van volwassen patiënten met secundaire progressieve multiple sclerose (SPMS) met actieve ziekte gedefinieerd door exacerbaties (relapses, schubs, opstoten) of kenmerken van ontstekingsactiviteit aangetoond door beeldvormende technieken (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
De behandeling met siponimod dient te worden gestart en gecontroleerd door een arts met ervaring op het gebied van multiple sclerose.
Voor de start van de behandeling moet bij patiënten het genotype voor CYP2C9 worden bepaald, om zo hun metaboliseerderstatus voor CYP2C9 te bepalen (zie rubriek 4.4, 4.5 en 5.2).
Bij patiënten met een CYP2C9*3*3‑genotype mag siponimod niet gebruikt worden (zie rubriek 4.3, 4.4 en 5.2).
Dosering
Start van de behandeling
De behandeling moet worden gestart met een titratieverpakking voor de eerste 5 dagen. De behandeling begint met 0,25 mg eenmaal daags op dag 1 en 2, gevolgd door eenmaaldaagse doses van 0,5 mg op dag 3, 0,75 mg op dag 4 en 1,25 mg op dag 5, om zo de aan de patiënt voorgeschreven onderhoudsdosis siponimod te bereiken, die start op dag 6 (zie Tabel 1).
Tijdens de eerste 6 dagen van de start van de behandeling dient de aanbevolen dagelijkse dosis eenmaal daags ’s ochtends te worden ingenomen met of zonder voedsel.
Tabel 1 Dosistitratieschema om de onderhoudsdosis te bereiken
Titratie | Titratiedosis | Titratieschema | Dosis |
Dag 1 | 0,25 mg | 1 x 0,25 mg | TITRATIE |
Dag 2 | 0,25 mg | 1 x 0,25 mg | |
Dag 3 | 0,5 mg | 2 x 0,25 mg | |
Dag 4 | 0,75 mg | 3 x 0,25 mg | |
Dag 5 | 1,25 mg | 5 x 0,25 mg | |
Dag 6 | 2 mg1 | 1 x 2 mg1 | ONDERHOUD |
1 Bij patiënten met een CYP2C9*2*3‑ of *1*3‑genotype is de aanbevolen onderhoudsdosis 1 mg eenmaal daags (1 x 1 mg of 4 x 0,25 mg) (zie hierboven en rubriek 4.4 en 5.2). Extra blootstelling van 0,25 mg op dag 5 vormt geen gevaar voor de veiligheid van de patiënt. | |||
Onderhoudsbehandeling
Bij patiënten met een CYP2C9*2*3‑ of *1*3‑genotype is de aanbevolen onderhoudsdosis 1 mg (zie rubriek 4.4 en 5.2).
De aanbevolen onderhoudsdosis van siponimod bij alle patiënten met een ander CYP2C9‑genotype is 2 mg.
Mayzent wordt eenmaal daags ingenomen.
Gemiste dosis tijdens de start van de behandeling
Als tijdens de eerste 6 dagen van de behandeling een titratiedosis wordt overgeslagen, moet de titratie opnieuw gestart worden met een nieuwe titratieverpakking.
Gemiste dosis na dag 6
Als een dosis wordt overgeslagen, dient de voorgeschreven dosis ingenomen te worden op het volgende geplande tijdstip. De volgende dosis dient niet verdubbeld te worden.
Herstart van de onderhoudsbehandeling na onderbreking van de behandeling
Als de onderhoudsbehandeling wordt onderbroken gedurende 4 of meer opeenvolgende dagelijkse doses, dient siponimod opnieuw gestart te worden met een nieuwe titratieverpakking.
Speciale patiëntengroepen
Ouderen
Siponimod is niet onderzocht bij patiënten van 65 jaar en ouder. Klinische onderzoeken includeerden patiënten tot en met 61 jaar. Voorzichtigheid is geboden bij het gebruik van siponimod bij ouderen vanwege onvoldoende gegevens over de veiligheid en werkzaamheid (zie rubriek 5.2).
Nierfunctiestoornis
Gebaseerd op klinisch-farmacologische studies is geen dosisaanpassing nodig bij patiënten met een nierfunctiestoornis (zie rubriek 5.2).
Leverfunctiestoornis
Siponimod mag niet worden gebruikt bij patiënten met een ernstige leverfunctiestoornis (Child-Pugh-klasse C) (zie rubriek 4.3). Alhoewel er geen dosisaanpassing nodig is bij patiënten met een lichte of matige leverfunctiestoornis, is voorzichtigheid geboden bij het initiëren van de behandeling bij deze patiënten (zie rubriek 4.4 en 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van siponimod bij kinderen en adolescenten in de leeftijd van 0 tot 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Oraal gebruik. Siponimod wordt met of zonder voedsel ingenomen.
De filmomhulde tabletten moeten in het geheel met water worden doorgeslikt.
4.3 Contra‑indicaties
- Overgevoeligheid voor de werkzame stof of voor pinda’s, soja of voor een van de in rubriek 6.1 vermelde hulpstoffen.
- Immunodeficiëntiesyndroom.
- Voorgeschiedenis van progressieve multifocale leuko-encefalopathie of cryptokokkenmeningitis.
- Actieve maligniteiten.
- Ernstige leverfunctiestoornis (Child-Pughklasse C).
- Patiënten die in de voorgaande 6 maanden een myocardinfarct (MI), instabiele angina pectoris, beroerte/transiënte ischemische aanval (TIA), gedecompenseerd hartfalen (waarvoor ziekenhuisopname vereist was), of New York Heart Association (NYHA)klasse III/IV hartfalen hebben gehad (zie rubriek 4.4).
- Patiënten met een voorgeschiedenis van tweedegraads atrioventriculair (AV) blok type Mobitz-II of derdegraads AVblok, sinoatriaal hartblok of sicksinussyndroom, als ze geen pacemaker dragen (zie rubriek 4.4).
- Patiënten die homozygoot zijn voor het CYP2C9*3 (CYP2C9*3*3)‑genotype (sterk vertraagde metaboliseerder).
- Tijdens de zwangerschap en bij vrouwen die zwanger kunnen worden en die geen effectieve anticonceptie gebruiken (zie rubriek 4.4 en 4.6).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Het veiligheidsprofiel van siponimod is gebaseerd op gegevens uit het kernonderdeel van de klinische studie. De meest voorkomende bijwerkingen die in het kernonderdeel van studie A2304 werden vastgesteld, waren hoofdpijn (15%) en hypertensie (12,6%). De veiligheidsgerelateerde informatie uit het verlengde deel van de langetermijnstudie A2304 was consistent met die waargenomen in het kernonderdeel.
Tabellarische lijst van bijwerkingen
Binnen elk(e) systeem/orgaanklasse worden bijwerkingen gerangschikt naar frequentie, met de meest voorkomende bijwerkingen eerst. Daarnaast is de overeenkomende frequentiecategorie voor elke bijwerking gebaseerd op de volgende afspraak: zeer vaak (≥1/10); vaak (≥1/100, <1/10); soms (≥1/1.000, <1/100); zelden (≥1/10.000, <1/1.000); zeer zelden (<1/10.000); niet bekend (kan met de beschikbare gegevens niet worden bepaald).
Tabel 2 Tabellarische lijst van bijwerkingen
Infecties en parasitaire aandoeningen | |
Vaak | Herpes zoster |
Zelden | Progressieve multifocale leuko-encefalopathie |
Niet bekend | Cryptokokkenhersenvliesontsteking |
Neoplasmata, benigne, maligne en niet‑gespecificeerd (inclusief cysten en poliepen) | |
Vaak | Naevus naevocellularis |
Soms | Plaveiselcelcarcinoom |
Bloed‑ en lymfestelselaandoeningen | |
Vaak | Lymfopenie |
Immuunsysteemaandoeningen | |
Zelden | Immuunreconstitutie-ontstekingssyndroom (Immune reconstitution inflammatory syndrome, IRIS) |
Zenuwstelselaandoeningen | |
Zeer vaak | Hoofdpijn |
Vaak | Duizeligheid |
Oogaandoeningen | |
Vaak | Macula‑oedeem |
Hartaandoeningen | |
Vaak | Bradycardie |
Bloedvataandoeningen | |
Zeer vaak | Hypertensie |
Maagdarmstelselaandoeningen | |
Vaak | Misselijkheid |
Skeletspierstelsel‑ en bindweefselaandoeningen | |
Vaak | Pijn in extremiteit |
Algemene aandoeningen en toedieningsplaatsstoornissen | |
Vaak | Perifeer oedeem |
Onderzoeken | |
Zeer vaak | Leverfunctietest verhoogd |
Vaak | Longfunctietest verlaagd |
Beschrijving van een selectie van de bijwerkingen
Infecties
In de klinische fase III‑studie bij SPMS-patiënten was de algehele infectiefrequentie bij patiënten die behandeld werden met siponimod vergelijkbaar met deze bij patiënten die placebo kregen (respectievelijk 49,0% versus 49,1%). Een hogere frequentie van infecties met herpes zoster werd echter gemeld bij het gebruik van siponimod (2,5%) in vergelijking met placebo (0,7%).
Gevallen van meningitis of meningo-encefalitis veroorzaakt door VZV zijn op elk moment tijdens de behandeling met siponimod opgetreden. Gevallen van CM zijn ook gemeld voor siponimod (zie rubriek 4.4).
Macula‑oedeem
Macula‑oedeem werd vaker gemeld bij patiënten die siponimod kregen (1,8%) dan bij patiënten die placebo kregen (0,2%). Hoewel het merendeel van de gevallen optrad binnen 3 tot 4 maanden na de start van siponimod, werden ook gevallen gemeld bij patiënten die langer dan 6 maanden werden behandeld met siponimod (zie rubriek 4.4). Enkele patiënten kregen last van een wazig zicht of verminderde gezichtsscherpte, maar anderen waren asymptomatisch en gediagnosticeerd bij routinematig oogheelkundig onderzoek. In het algemeen verbeterde of verdween het macula‑oedeem spontaan na het staken van de behandeling. Het risico op herhaling na hernieuwde blootstelling is niet onderzocht.
Bradyaritmie
Het initiëren van een behandeling met siponimod resulteert in een tijdelijke afname van de hartslag en kan ook gepaard gaan met vertragingen van de atrioventriculaire geleiding (zie rubriek 4.4). Bradycardie werd gemeld bij 6,2% van de patiënten die behandeld werden met siponimod in vergelijking met 3,1% van de patiënten die placebo kregen. AV‑blok werd gemeld bij 1,7% van de patiënten die behandeld werden met siponimod in vergelijking met 0,7% van de patiënten die placebo kregen (zie rubriek 4.4).
De maximale afname van de hartslag wordt gezien in de eerste 6 uur na toediening van de dosis.
Een tijdelijke, dosisafhankelijke verlaging van de hartslag werd gezien tijdens de eerste toedieningsfase en stabiliseerde bij doses ≥5 mg. Bradyaritmische voorvallen (AV‑blokken en sinuspauzes) werden met een hogere incidentie waargenomen bij de behandeling met siponimod in vergelijking met placebo.
De meeste AV‑blokken en sinuspauzes traden op bij doses hoger dan de therapeutische dosis van 2 mg. Een beduidend hogere incidentie werd waargenomen wanneer er niet getitreerd werd dan wanneer er wel sprake was van dosistitratie.
De afname van de hartslag die wordt geïnduceerd door siponimod kan worden tenietgedaan door toediening van atropine of isoprenaline.
Leverfunctietesten
Verhoogde leverenzymen (voornamelijk ALAT‑verhoging) zijn gemeld bij MS‑patiënten die met siponimod werden behandeld. In de fase III‑studie bij SPMS-patiënten werden verhogingen van de leverfunctietesten vaker gezien bij patiënten die siponimod gebruikten (11,3%) dan bij patiënten die placebo kregen (3,1%). Dit is vooral te wijten aan verhogingen van levertransaminasen (ALAT/ASAT) en gammaglutamyltransferase (GGT). Het merendeel van de verhogingen trad op binnen 6 maanden na de start van de behandeling. ALAT‑waarden keerden terug naar normaalwaarden binnen ongeveer 1 maand na het staken van siponimod (zie rubriek 4.4).
Bloeddruk
In de klinische fase III‑studie met SPMS werd hypertensie vaker gemeld bij patiënten die siponimod kregen (12,6%) dan bij patiënten die placebo kregen (9,0%). De behandeling met siponimod leidde tot een stijging van de systolische en diastolische bloeddruk. Deze stijging begon snel na de start van de behandeling en het maximale effect werd bereikt na ongeveer 6 maanden behandeling (systolisch 3 mmHg, diastolisch 1,2 mmHg) en bleef hierna stabiel. Het effect hield aan bij voortgezette behandeling.
Convulsies
In de klinische fase III‑studie bij SPMS-patiënten werden convulsies gemeld bij 1,7% van de patiënten die behandeld werden met siponimod in vergelijking met 0,4% van de patiënten die placebo kregen.
Effecten op het ademhalingsstelsel
Kleine verlagingen in de waarden van het geforceerd expiratoir volume binnen 1 seconde (FEV1) en in de diffusiecapaciteit van de longen voor koolstofmonoxide (DLCO) werden waargenomen bij de behandeling met siponimod. In de klinische fase III‑studie bij SPMS-patiënten waren op maand 3 en 6 van de behandeling de gemiddelde veranderingen ten opzichte van baseline in FEV1 in de siponimod‑groep ‑0,1 l op elk tijdstip, ten opzichte van geen verandering in de placebogroep. Deze observaties waren iets hoger (ongeveer 0,15 l gemiddelde verandering ten opzichte van baseline in FEV1) bij patiënten met ademhalingsstoornissen zoals chronisch obstructieve longziekte (COPD) of astma die werden behandeld met siponimod. Bij chronische behandeling veroorzaakte deze verlaging geen klinisch significante bijwerkingen en was deze niet gerelateerd aan een toename van het aantal meldingen van hoesten of dyspneu (zie rubriek 5.1).
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be

7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Ierland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Mayzent 0,25 mg filmomhulde tabletten
EU/1/19/1414/001
EU/1/19/1414/002
EU/1/19/1414/004
Mayzent 1 mg filmomhulde tabletten
EU/1/19/1414/007
EU/1/19/1414/008
Mayzent 2 mg filmomhulde tabletten
EU/1/19/1414/003
EU/1/19/1414/005
EU/1/19/1414/006
10. DATUM VAN HERZIENING VAN DE TEKST
06.05.2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4166732 | MAYZENT 0,25MG FILMOMH TABL 12 | L04AA42 | € 278,92 | € 244,45 | Ja | € 12,8 | € 8,5 |
| 4166724 | MAYZENT 0,25MG FILMOMH TABL 120 | L04AA42 | € 1616,1 | € 1466,79 | Ja | - | - |
| 4166716 | MAYZENT 2,00MG FILMOMH TABL 28 | L04AA42 | € 1511,6 | € 1369 | Ja | € 12,8 | € 8,5 |
| 4506754 | MAYZENT 1,00MG FILMOMH TABL 28 | L04AA42 | € 1511,6 | - | Ja | € 12,8 | € 8,5 |