. NAAM VAN HET GENEESMIDDEL
Kesimpta 20 mg oplossing voor injectie in een voorgevulde spuit
Kesimpta 20 mg oplossing voor injectie in een voorgevulde pen
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Kesimpta 20 mg oplossing voor injectie in een voorgevulde spuit
Elke voorgevulde spuit bevat 20 mg ofatumumab in 0,4 ml oplossing (50 mg/ml).
Kesimpta 20 mg oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen bevat 20 mg ofatumumab in 0,4 ml oplossing (50 mg/ml).
Ofatumumab is een volledig humaan monoklonaal antilichaam dat in een muriene cellijn (NS0) wordt geproduceerd met behulp van DNA-recombinatietechniek.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie (injectievloeistof)
Oplossing voor injectie (injectievloeistof) in een voorgevulde pen (Sensoready-pen)
De oplossing is helder tot bijna doorschijnend en kleurloos tot licht bruingeel.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Kesimpta is geïndiceerd voor de behandeling van volwassen patiënten met relapsing multiple sclerose (RMS) met actieve ziekte gedefinieerd door klinische of beeldvormende technieken (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
De behandeling dient te worden opgestart door een arts met ervaring in de behandeling van neurologische aandoeningen.
Dosering
De aanbevolen dosis is 20 mg ofatumumab toegediend door middel van subcutane injectie met:
- initiële doses in week 0, 1 en 2, gevolgd door
- maandelijkse onderhoudsdoses vanaf week 4.
Gemiste doses
Als een injectie wordt gemist, moet de toediening zo spoedig mogelijk plaatsvinden, zonder te wachten tot de volgende geplande dosis. De daaropvolgende doses moeten worden toegediend volgens de aanbevolen intervallen.
Bijzondere patiëntengroepen
Volwassenen ouder dan 55 jaar
Er zijn geen studies uitgevoerd bij MS-patiënten die ouder zijn dan 55 jaar. Op basis van de beperkte beschikbare gegevens wordt een dosisaanpassing niet noodzakelijk geacht bij patiënten ouder dan 55 jaar (zie rubriek 5.2).
Verminderde nierfunctie
Bij patiënten met een verminderde nierfunctie is naar verwachting geen aanpassing van de dosis nodig (zie rubriek 5.2).
Verminderde leverfunctie
Bij patiënten met een verminderde leverfunctie is naar verwachting geen aanpassing van de dosis nodig (zie rubriek 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van Kesimpta bij kinderen in de leeftijd van 0 tot 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Dit geneesmiddel is bedoeld voor zelftoediening door de patiënt door middel van subcutane injectie.
De gebruikelijke plaatsen voor subcutane injecties zijn de buik, de bovenbenen en de buitenzijde van de bovenarm.
De eerste injectie dient onder begeleiding van een beroepsbeoefenaar in de gezondheidszorg te worden uitgevoerd (zie rubriek 4.4).
De bijsluiter bevat uitgebreide instructies voor de toediening.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Patiënten in een ernstig immuungecompromitteerde toestand (zie rubriek 4.4).
Ernstige actieve infectie totdat deze infectie is verdwenen (zie rubriek 4.4).
Patiënten met een bevestigde actieve maligniteit.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De belangrijkste en vaakst gemelde bijwerkingen zijn bovensteluchtweginfecties (39,4%), systemische injectiegerelateerde reacties (20,6%), lokale reacties op de injectieplaats (10,9%) en urineweginfecties (11,9%) (zie rubriek 4.4 en onderstaand subrubriek “Beschrijving van enkele specifieke bijwerkingen” voor meer informatie).
Lijst van bijwerkingen in tabelvorm
Bijwerkingen die in verband met het gebruik van ofatumumab in klinische RMS-hoofdstudies en uit ervaringen in de post-marketingfase zijn gemeld, staan in tabel 1 vermeld naar systeem/orgaanklasse volgens MedDRA. Binnen elke systeem/orgaanklasse worden de bijwerkingen gerangschikt in volgorde van frequentie, met de vaakst voorkomende bijwerkingen eerst. Binnen elke frequentiegroep zijn de bijwerkingen gerangschikt volgens afnemende ernst. Daarnaast is de overeenkomstige frequentiecategorie voor elke bijwerking gebaseerd op de volgende afspraak: zeer vaak (≥1/10); vaak (≥1/100, <1/10); soms (≥1/1.000, <1/100); zelden (≥1/10.000, <1/1.000); zeer zelden (<1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald).
Tabel 1 Lijst van bijwerkingen in tabelvorm
Infecties en parasitaire aandoeningen | |
Zeer vaak | Infecties van bovenste luchtwegen1 |
Vaak | Orale herpes |
Immuunsysteemaandoeningen | |
Niet bekend | Overgevoeligheidsreacties3 |
Maagdarmstelselaandoeningen | |
Vaak | Nausea, braken4 |
Algemene aandoeningen en toedieningsplaatsstoornissen | |
Zeer vaak | Injectieplaatsreacties (lokaal) |
Onderzoeken | |
Vaak | Bloed immunoglobuline M verlaagd |
Letsels, intoxicaties en verrichtingscomplicaties | |
Zeer vaak | Injectiegerelateerde reacties (systemisch) |
1 Groepering van voorkeurstermen (preferred terms, PT’s) is overwogen voor het bepalen van de frequentie van bijwerkingen en omvat het volgende: nasofaryngitis, infectie van bovenste luchtwegen, griep, sinusitis, faryngitis, rhinitis, virale bovensteluchtweginfectie, tonsillitis, acute sinusitis, faryngotonsillitis, laryngitis, streptokokkenfaryngitis, virale rhinitis, bacteriële sinusitis, bacteriële tonsillitis, virale faryngitis, virale tonsillitis, chronische sinusitis, nasale herpes, tracheïtis. | |
Beschrijving van geselecteerde bijwerkingen
Infecties
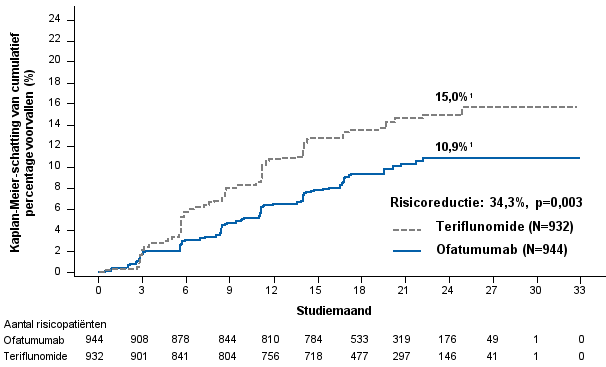
In de klinische fase III-studies naar RMS, was het totaal aantal infecties en ernstige infecties bij patiënten behandeld met ofatumumab vergelijkbaar met patiënten die behandeld werden met teriflunomide (respectievelijk 51,6% vs. 52,7% en 2,5% vs. 1,8%). Twee patiënten (0,2%) stopten met de studie en 11 patiënten (1,2%) onderbraken tijdelijk de studiebehandeling vanwege een ernstige infectie.
Bovensteluchtweginfecties
In deze studies had 39,4% van de patiënten behandeld met ofatumumab bovensteluchtweginfecties in vergelijking met 37,8% van de patiënten behandeld met teriflunomide. De infecties waren voornamelijk licht tot matig en bestonden voor het merendeel uit nasofaryngitis, bovenste luchtweginfectie en griep.
Systemische injectiegerelateerde reacties
In de klinische fase III-studies naar RMS werden SIRR’s (systemische injectiegerelateerde reacties [Systemic injection-related reactions]) gemeld bij 20,6% van de patiënten behandeld met ofatumumab.
De incidentie van SIRR’s was het hoogst bij de eerste injectie (14,4%) en nam significant af bij daaropvolgende injecties (4,4% bij de tweede injectie en < 3% bij de derde). De SIRR’s waren in de meeste gevallen (99,8%) licht tot matig qua ernst. Twee (0,2%) met ofatumumab behandelde MS-patiënten meldden ernstige injectiegerelateerde reacties, maar deze waren niet levensbedreigend. De vaakst gemelde symptomen (≥ 2%) waren onder andere koorts, hoofdpijn, myalgie, koude rillingen en vermoeidheid. Bijkomende gemelde symptomen waren nausea (1,7%) en braken (0,6%).
Lokale reacties op de injectieplaats
In de klinische fase III-studies naar RMS werden lokale reacties op de injectieplaats gemeld bij 10,9% van de patiënten behandeld met ofatumumab.
Lokale reacties op de injectieplaats deden zich zeer vaak voor. Deze waren alle van een lichte tot matige graad en niet-ernstig van aard. De vaakst gemelde symptomen (≥ 2%) waren onder andere erytheem, pijn, jeuk en zwelling.
Laboratoriumafwijkingen
Immunoglobulinen
In de loop van de klinische fase III-studies naar RMS werd een daling van de gemiddelde immunoglobuline M (IgM)-waarde waargenomen (daling van 30,9% na 48 weken en 38,8% na 96 weken), maar er kon geen verband worden aangetoond tussen deze daling en het risico op infecties, waaronder ernstige infecties.
Bij 14,3% van de patiënten resulteerde de behandeling met ofatumumab in een daling van de IgM-waarde tot onder 0,34 g/l.
Ofatumumab werd geassocieerd met een tijdelijke daling met 4,3% van de gemiddelde immunoglobuline G (IgG)-spiegel na 48 weken behandeling, maar met een stijging met 2,2% na 96 weken.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Ierland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/21/1532/001‑004
10. DATUM VAN HERZIENING VAN DE TEKST
09/01/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4349932 | KESIMPTA 20MG SOL INJ VOORGEV.PEN 1 (SENSOREADY) | L04AA52 | € 1699,51 | - | Ja | € 12,8 | € 8,5 |