BIJLAGE I
SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
RINVOQ 15 mg tabletten met verlengde afgifte
RINVOQ 30 mg tabletten met verlengde afgifte
RINVOQ 45 mg tabletten met verlengde afgifte
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
RINVOQ 15 mg, tabletten met verlengde afgifte
Elke tablet met verlengde afgifte bevat upadacitinib-hemihydraat, overeenkomend met 15 mg upadacitinib.
RINVOQ 30 mg, tabletten met verlengde afgifte
Elke tablet met verlengde afgifte bevat upadacitinib-hemihydraat, overeenkomend met 30 mg upadacitinib.
RINVOQ 45 mg, tabletten met verlengde afgifte
Elke tablet met verlengde afgifte bevat upadacitinib-hemihydraat, overeenkomend met 45 mg upadacitinib.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Tablet met verlengde afgifte
RINVOQ 15 mg, tabletten met verlengde afgifte
Paarse, 14 x 8 mm, langwerpige, biconvexe tablet met verlengde afgifte met aan één zijde de opdruk 'a15'.
RINVOQ 30 mg, tabletten met verlengde afgifte
Rode, 14 x 8 mm, langwerpige, biconvexe tablet met verlengde afgifte met aan één zijde de opdruk ‘a30’.
RINVOQ 45 mg, tabletten met verlengde afgifte
Gele tot gespikkeld gele, 14 x 8 mm, langwerpige, biconvexe tablet met verlengde afgifte met aan één zijde de opdruk ‘a45’.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Reumatoïde artritis
RINVOQ is geïndiceerd voor de behandeling van matig tot ernstig actieve reumatoïde artritis bij volwassen patiënten die onvoldoende hebben gereageerd op een of meer disease-modifying antirheumatic drugs (DMARD's) of die niet kunnen verdragen. RINVOQ kan worden gebruikt als monotherapie of in combinatie met methotrexaat.
Artritis psoriatica
RINVOQ is geïndiceerd voor de behandeling van actieve artritis psoriatica bij volwassen patiënten die onvoldoende hebben gereageerd op een of meer DMARD's of die niet kunnen verdragen. RINVOQ kan worden gebruikt als monotherapie of in combinatie met methotrexaat.
Axiale spondyloartritis
Niet-radiografische axiale spondyloartritis (nr-axSpA)
RINVOQ is geïndiceerd voor de behandeling van actieve, niet-radiografische axiale spondyloartritis bij volwassen patiënten met objectieve tekenen van ontsteking, zoals een verhoogd C-reactief proteïne (CRP) en/of magnetic resonance imaging (MRI), die onvoldoende hebben gereageerd op niet-steroïde ontstekingsremmers (NSAID’s).
Spondylitis ankylopoetica (AS, radiografische axiale spondyloartritis)
RINVOQ is geïndiceerd voor de behandeling van actieve spondylitis ankylopoetica bij volwassen patiënten die onvoldoende hebben gereageerd op conventionele behandeling.
Reuscelarteriitis
RINVOQ is geïndiceerd voor de behandeling van reuscelarteriitis bij volwassen patiënten.
Atopische dermatitis
RINVOQ is geïndiceerd voor de behandeling van matige tot ernstige atopische dermatitis (constitutioneel eczeem) bij volwassenen en adolescenten vanaf 12 jaar die in aanmerking komen voor systemische therapie.
Alopecia areata
RINVOQ is geïndiceerd voor de behandeling van ernstige alopecia areata bij volwassenen en adolescenten vanaf 12 jaar (zie rubriek 5.1).
Vitiligo
RINVOQ is geïndiceerd voor de behandeling van non-segmentale vitiligo bij volwassenen en adolescenten vanaf 12 jaar die in aanmerking komen voor systemische therapie.
Colitis ulcerosa
RINVOQ is geïndiceerd voor de behandeling van volwassen patiënten met matig tot ernstig actieve colitis ulcerosa (CU) die onvoldoende reageerden op, niet meer reageerden op of intolerant waren voor een conventionele behandeling of een biologisch geneesmiddel (‘biological’).
Ziekte van Crohn
RINVOQ is geïndiceerd voor de behandeling van volwassen patiënten met matig tot ernstig actieve ziekte van Crohn die onvoldoende reageerden op, niet meer reageerden op of intolerant waren voor een conventionele behandeling of een biologisch geneesmiddel (‘biological’).
4.2 Dosering en wijze van toediening
Een behandeling met upadacitinib moet worden gestart en plaatsvinden onder toezicht van artsen die ervaring hebben met de diagnose en behandeling van aandoeningen waarvoor upadacitinib is geïndiceerd.
Dosering
Reumatoïde artritis, artritis psoriatica en axiale spondyloartritis
De aanbevolen dosis upadacitinib is 15 mg eenmaal daags.
Als patiënten met axiale spondyloartritis na 16 weken behandeling geen klinische respons hebben, dient stopzetten van de therapie te worden overwogen. Bij sommige patiënten met aanvankelijk een gedeeltelijke respons kan de respons later nog verbeteren bij voortzetting van de behandeling na 16 weken.
Reuscelarteriitis
De aanbevolen dosis upadacitinib is 15 mg eenmaal daags in combinatie met een afbouwende corticosteroïdenkuur. Monotherapie met upadacitinib mag niet worden gebruikt voor de behandeling van acute recidieven (zie rubriek 4.4).
Op basis van de chronische aard van reuscelarteriitis kan na stopzetting van de behandeling met corticosteroïden de behandeling met upadacitinib 15 mg eenmaal daags worden voortgezet als monotherapie.
Atopische dermatitis
De aanbevolen dosis upadacitinib is 15 mg of 30 mg eenmaal daags, afhankelijk van de toestand van de individuele patiënt:
- Een dosis van 15 mg is aanbevolen voor patiënten die een hoger risico hebben op veneuze trombo-embolie (VTE), een ernstig ongewenst cardiovasculair voorval (MACE; major adverse cardiovascular event) en een maligniteit (zie rubriek 4.4).
- Een dosis van 30 mg eenmaal daags kan passend zijn voor patiënten met een hoge ziektelast die geen hoger risico hebben op VTE, een MACE en een maligniteit (zie rubriek 4.4) of voor patiënten met onvoldoende respons op 15 mg eenmaal daags.
- Voor adolescenten (van 12 tot en met 17 jaar) met een gewicht van ten minste 30 kg wordt een dosering van 15 mg aanbevolen. Als de patiënt niet adequaat reageert op 15 mg eenmaal daags, kan de dosering verhoogd worden tot 30 mg eenmaal daags.
- De laagste effectieve dosis die nodig is voor aanhouden van de respons moet worden gebruikt.
Bij patiënten van 65 jaar en ouder is de aanbevolen dosis 15 mg eenmaal daags (zie rubriek 4.4).
Gelijktijdige topische therapieën
Upadacitinib kan worden gebruikt met of zonder topische corticosteroïden. Topische calcineurineremmers mogen worden gebruikt voor gevoelige gebieden zoals het gezicht, de hals en intertrigineuze en genitale gebieden.
Bij patiënten die na 12 weken behandeling geen tekenen vertonen van therapeutisch voordeel, moet staken van de behandeling met upadacitinib worden overwogen.
Alopecia areata
Voor volwassen en adolescente patiënten vanaf 12 jaar met een gewicht van ten minste 30 kg is de aanbevolen dosis upadacitinib eenmaal daags 15 mg of 30 mg, afhankelijk van de toestand van de individuele patiënt.
- Een dosis van 30 mg eenmaal daags kan passend zijn voor patiënten met een hoge ziektelast of patiënten met onvoldoende respons op 15 mg eenmaal daags.
- De laagste effectieve dosis die nodig is voor aanhouden van de respons moet worden gebruikt.
Bij patiënten van 65 jaar en ouder is de aanbevolen dosis eenmaal daags 15 mg (zie rubriek 4.4).
Werkzaamheidsgegevens zijn beschikbaar voor een behandelperiode van 24 weken. De voordelen en risico’s van de behandeling dienen op gezette tijden individueel te worden herbeoordeeld.
Vitiligo
De aanbevolen dosis upadacitinib is 15 mg eenmaal daags voor volwassenen en adolescenten vanaf 12 jaar en ouder die ten minste 30 kg wegen.
Bij patiënten die na 48 weken behandeling geen tekenen vertonen van therapeutisch voordeel, moet staken van de behandeling met upadacitinib worden overwogen.
Colitis ulcerosa
Inductie
De aanbevolen inductiedosis upadacitinib is 45 mg eenmaal daags gedurende 8 weken. Bij patiënten die na 8 weken geen voldoende therapeutisch voordeel hebben bereikt, kan de dosis van 45 mg upadacitinib eenmaal daags voor een periode van nog eens 8 weken worden voortgezet (zie rubriek 5.1). Bij patiënten die na week 16 geen bewijs van therapeutisch voordeel vertonen, moet de behandeling met upadacitinib worden stopgezet.
Onderhoud
De aanbevolen onderhoudsdosis upadacitinib is 15 mg of 30 mg eenmaal daags, afhankelijk van de toestand van de individuele patiënt:
- Een dosis van 15 mg is aanbevolen voor patiënten die een hoger risico hebben op VTE, MACE en maligniteiten (zie rubriek 4.4).
- Voor sommige patiënten kan een dosis van 30 mg eenmaal daags passend zijn, bijvoorbeeld voor patiënten met een hoge ziektelast of patiënten bij wie een inductiebehandeling van 16 weken nodig is, maar die geen hoger risico hebben op VTE, MACE en maligniteiten (zie rubriek 4.4), of voor patiënten die bij een dosis van 15 mg eenmaal daags niet voldoende baat hebben bij de behandeling.
- De laagste effectieve dosis die nodig is voor aanhouden van de respons moet worden gebruikt.
Bij patiënten van 65 jaar en ouder is de aanbevolen dosis 15 mg eenmaal daags (zie rubriek 4.4).
Bij patiënten die respons vertonen op de behandeling met upadacitinib, kan de behandeling met corticosteroïden worden gereduceerd en/of stopgezet in overeenstemming met de zorgstandaard.
Ziekte van Crohn
Inductie
De aanbevolen inductiedosis upadacitinib is 45 mg eenmaal daags gedurende 12 weken. Bij patiënten die na de aanvankelijke inductie gedurende 12 weken geen voldoende therapeutisch voordeel hebben bereikt, kan overwogen worden de inductie met nog eens 12 weken te verlengen met een dosis van 30 mg eenmaal daags. Bij deze patiënten moet de behandeling met upadacitinib worden stopgezet als er na een behandeling van 24 weken geen bewijs van therapeutisch voordeel is.
Onderhoud
De aanbevolen onderhoudsdosis upadacitinib is 15 mg of 30 mg eenmaal daags, afhankelijk van de toestand van de individuele patiënt:
- Een dosis van 15 mg is aanbevolen voor patiënten die een hoger risico hebben op VTE, MACE en maligniteiten (zie rubriek 4.4).
- Een dosis van 30 mg eenmaal daags kan passend zijn voor patiënten met een hoge ziektelast, maar zonder hoger risico op VTE, MACE en maligniteiten (zie rubriek 4.4), of voor patiënten die bij een dosis van 15 mg eenmaal daags niet voldoende baat hebben.
- De laagste effectieve dosis die nodig is voor aanhouden van de respons moet worden gebruikt.
Bij patiënten van 65 jaar en ouder is de aanbevolen onderhoudsdosis 15 mg eenmaal daags (zie rubriek 4.4).
Bij patiënten die respons vertonen op de behandeling met upadacitinib, kan de behandeling met corticosteroïden worden gereduceerd en/of stopgezet in overeenstemming met de zorgstandaard.
Interacties
Bij patiënten met colitis ulcerosa en de ziekte van Crohn die een sterke cytochroom P450 (CYP) 3A4-remmer krijgen (bijvoorbeeld ketoconazol of claritromycine), is de aanbevolen inductiedosis 30 mg eenmaal daags en de aanbevolen onderhoudsdosis 15 mg eenmaal daags (zie rubriek 4.5).
Starten van de behandeling
Een behandeling mag niet worden ingezet bij patiënten met een absolute lymfocytentelling (ALC) < 0,5 x 109 cellen/l, een absolute neutrofielentelling (ANC) < 1 x 109 cellen/l of een hemoglobinewaarde (Hb) < 8 g/dl (zie rubriek 4.4 en 4.8).
Onderbreking van behandeling
De behandeling moet worden onderbroken als een patiënt een ernstige infectie krijgt tot de infectie onder controle is.
Onderbreking van de behandeling kan nodig zijn voor het herstel van afwijkende laboratoriumwaarden, zoals beschreven in tabel 1.
Tabel 1 Laboratoriumwaarden en richtlijn voor monitoring
Laboratoriumwaarde | Actie | Richtlijn voor monitoring |
Absolute neutrofielentelling (ANC) | De behandeling moet worden onderbroken als ANC < 1 x 109 cellen/l is en mag worden hervat wanneer ANC boven deze waarde is teruggekeerd | Evalueer bij baseline en daarna niet later dan 12 weken na start van de behandeling. Evalueer vervolgens tijdens de individuele controles van de patiënt. |
Absolute lymfocytentelling (ALC) | De behandeling moet worden onderbroken als ALC < 0,5 x 109 cellen/l is en mag worden hervat wanneer ALC boven deze waarde is teruggekeerd | |
Hemoglobine (Hb) | De behandeling moet worden onderbroken als de Hb < 8 g/dl is en mag worden hervat wanneer de Hb boven deze waarde is teruggekeerd | |
Levertransaminasen | De behandeling moet tijdelijk worden onderbroken bij een vermoeden van door geneesmiddelen geïnduceerd leverletsel | Evalueer bij baseline en daarna bij routinecontroles van de patiënt. |
Lipiden | Patiënten moeten worden behandeld volgens de internationale klinische richtlijnen voor hyperlipidemie | Evalueer 12 weken na aanvang van de behandeling en daarna volgens de internationale klinische richtlijnen voor hyperlipidemie |
Speciale patiëntengroepen
Ouderen
Reumatoïde artritis, artritis psoriatica en axiale spondyloartritis
Er zijn beperkte gegevens voor patiënten van 75 jaar en ouder (zie rubriek 4.4).
Atopische dermatitis
Bij atopische dermatitis worden doses hoger dan 15 mg eenmaal daags niet aanbevolen voor patiënten van 65 jaar en ouder (zie rubriek 4.4 en 4.8).
Alopecia areata
De veiligheid en werkzaamheid van upadacitinib bij patiënten van 65 jaar en ouder zijn nog niet vastgesteld (zie rubriek 4.4).
Vitiligo
Er zijn beperkte gegevens voor patiënten van 65 jaar en ouder (zie rubriek 4.4 en 4.8).
Colitis ulcerosa en ziekte van Crohn
Bij colitis ulcerosa en de ziekte van Crohn worden doses hoger dan 15 mg eenmaal daags als onderhoudsbehandeling niet aanbevolen voor patiënten van 65 jaar en ouder (zie rubriek 4.4 en 4.8). De veiligheid en werkzaamheid van upadacitinib bij patiënten van 75 jaar en ouder zijn nog niet vastgesteld.
Nierfunctiestoornis
Er is geen dosisaanpassing nodig voor patiënten met een lichte of matige nierfunctiestoornis. Er zijn beperkte gegevens over het gebruik van upadacitinib bij patiënten met een ernstige nierfunctiestoornis (zie rubriek 5.2). Upadacitinib moet voorzichtig worden gebruikt bij patiënten met ernstige nierfunctiestoornissen zoals beschreven in tabel 2. Het gebruik van upadacitinib is nog niet onderzocht bij patiënten met eindstadium nierfalen en wordt daarom niet aanbevolen voor gebruik bij deze patiënten.
Tabel 2 Aanbevolen dosis bij een ernstige nierfunctiestoornis a
Therapeutische indicatie | Aanbevolen dosis eenmaal daags |
Reumatoïde artritis, artritis psoriatica, axiale spondyloartritis, reuscelarteriitis, atopische dermatitis, alopecia areata en vitiligo | 15 mg |
Colitis ulcerosa, ziekte van Crohn | Inductie: 30 mg |
Onderhoud: 15 mg | |
a geschatte glomerulaire filtratiesnelheid (eGFR) 15 tot < 30 ml/min/1,73 m2 | |
Leverfunctiestoornis
Er is geen dosisaanpassing nodig voor patiënten met lichte (Child‑Pugh A) of matige (Child‑Pugh B) leverfunctiestoornis (zie rubriek 5.2). Upadacitinib mag niet worden gebruikt bij patiënten met ernstige (Child‑Pugh C) leverfunctiestoornis (zie rubriek 4.3).
Pediatrische patiënten
De veiligheid en werkzaamheid van RINVOQ bij kinderen jonger dan 12 jaar met atopische dermatitis, alopecia areata of vitiligo zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
De veiligheid en werkzaamheid van RINVOQ bij kinderen en jongeren met reumatoïde artritis, artritis psoriatica, axiale spondyloartritis, colitis ulcerosa of de ziekte van Crohn in de leeftijd van 0 tot 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Er is geen relevante toepassing van RINVOQ bij pediatrische patiënten voor de indicatie reuscelarteriitis.
Wijze van toediening
RINVOQ moet eenmaal per dag oraal met of zonder voedsel worden ingenomen en mag op elk moment van de dag worden ingenomen. De tabletten moeten in hun geheel worden doorgeslikt en mogen niet worden gesplitst of fijngemaakt en er mag niet op gekauwd worden om er zeker van te zijn dat de gehele dosis correct wordt ingenomen.
4.3 Contra-indicaties
- Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
- Actieve tuberculose (tb) of actieve ernstige infecties (zie rubriek 4.4).
- Ernstige leverfunctiestoornis (zie rubriek 4.2).
- Zwangerschap (zie rubriek 4.6).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Bij de placebogecontroleerde klinische onderzoeken naar reumatoïde artritis, artritis psoriatica en axiale spondyloartritis waren de meest gemelde bijwerkingen (≥ 2% van de patiënten bij ten minste één van de indicaties met het hoogste percentage onder de genoemde indicaties) met 15 mg upadacitinib: infecties van de bovenste luchtwegen (19,5%), verhoogde creatinekinase (CK) in het bloed (8,6%), verhoogde alanineaminotransferase (4,3%) bronchitis (3,9%), misselijkheid (3,5%), neutropenie (2,8%), hoesten (2,2%), verhoogde aspartaataminotransferase (2,2%) en hypercholesterolemie (2,2%).
Bij de placebogecontroleerde klinische onderzoeken naar atopische dermatitis waren de meest gemelde bijwerkingen (≥ 2% van de patiënten) met 15 mg of 30 mg upadacitinib infectie van de bovenste luchtwegen (25,4%), acne (15,1%), herpes simplex (8,4%), hoofdpijn (6,3%), verhoogde CK in het bloed (5,5%), hoesten (3,2%), folliculitis (3,2%), buikpijn (2,9%), misselijkheid (2,7%), neutropenie (2,3%), pyrexie (2,1%) en influenza (2,1%).
In de placebogecontroleerde klinische onderzoeken naar de inductie- en onderhoudsdosis voor colitis ulcerosa en de ziekte van Crohn waren de meest gemelde bijwerkingen (≥ 3% van de patiënten) bij 45 mg, 30 mg of 15 mg upadacitinib een bovensteluchtweginfectie (19,9%), pyrexie (8,7%), verhoogde creatinekinase (CK) in het bloed (7,6%), anemie (7,4%), hoofdpijn (6,6%), acne (6,3%), herpes zoster (6,1%), neutropenie (6,0%), rash (5,2%), pneumonie (4,1%), hypercholesterolemie (4,0%), bronchitis (3,9%), verhoogde aspartaataminotransferase (3,9%), vermoeidheid (3,9%), folliculitis (3,6%), verhoogde alanineaminotransferase (3,5%), herpes simplex (3,2%) en influenza (3,2%).
De meest gemelde ernstige bijwerkingen waren ernstige infecties (zie rubriek 4.4).
Het veiligheidsprofiel van upadacitinib bij langdurige behandeling was over het algemeen vergelijkbaar met het veiligheidsprofiel tijdens de placebogecontroleerde periode bij alle indicaties.
Lijst van bijwerkingen in tabelvorm
De onderstaande lijst met bijwerkingen is gebaseerd op ervaringen van klinische onderzoeken en postmarketingervaring. De frequentie van bijwerkingen die hieronder worden vermeld, wordt gedefinieerd aan de hand van de volgende conventie: zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000). De frequenties in tabel 3 zijn gebaseerd op de hogere aantallen bijwerkingen die werden gemeld bij RINVOQ in klinisch onderzoek naar reumatologische aandoeningen (15 mg), atopische dermatitis (15 mg en 30 mg), alopecia areata (15 mg en 30 mg), vitiligo (15 mg), colitis ulcerosa (15 mg, 30 mg en 45 mg) of de ziekte van Crohn (15 mg, 30 mg en 45 mg). In het geval van merkbare verschillen in frequentie tussen de indicaties wordt dit aangegeven in de voetnoten onder de tabel.
Tabel 3 Bijwerkingen
Systeem/orgaanklasse | Zeer vaak | Vaak | Soms | Zelden |
Infecties en parasitaire aandoeningen | Bovenste-luchtweginfectiesa | Bronchitisa,b | Orale candidiase |
|
Neoplasmata, benigne, maligne en niet-gespecificeerd (inclusief cysten en poliepen) |
| Niet-melanoom huidkankerf |
|
|
Bloed‑ en lymfestelselaandoeningen |
| Anemiea |
|
|
Immuunsysteemaandoeningen |
| Urticariac,g | Ernstige overgevoelig-heidsreactiesa,e |
|
Voedings- en stofwisselingsstoornissen |
| Hyperchole-sterolemiea,b | Hypertri-glyceridemie |
|
Zenuwstelselaandoeningen |
| Hoofdpijna,j |
|
|
Evenwichtsorgaan- en ooraandoeningen |
| Vertigoa |
|
|
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen |
| Hoesten |
|
|
Maagdarmstelselaandoeningen |
| Buikpijna | Gastro‑intesti-nale perforatiei |
|
Huid- en onderhuidaandoeningen | Acnea,c,d,g | Rasha |
|
|
Voortplantingsstelsel- en borstaandoeningen |
|
|
| Sperma-verkleuringl |
Algemene aandoeningen en toedieningsplaatsstoornissen |
| Vermoeidheid |
|
|
Onderzoeken |
| Verhoogde creatinekinase (CK) in bloed |
|
|
a Weergegeven als gegroepeerde term | ||||
Beschrijving van geselecteerde bijwerkingen
Reumatoïde artritis
Infecties
In placebogecontroleerde klinische onderzoeken met achtergrond-DMARD's, was de frequentie van infecties over een periode van 12/14 weken in de groep die 15 mg upadacitinib kreeg 27,4% vergeleken met 20,9% in de placebogroep. In methotrexaat (MTX)-gecontroleerde klinische onderzoeken was de frequentie van infecties over een periode van 12/14 weken in de groep die monotherapie met 15 mg upadacitinib kreeg 19,5% vergeleken met 24,0% in de MTX-groep. De algehele ratio infecties op lange termijn voor de groep die 15 mg upadacitinib kreeg over alle vijf klinische fase 3-onderzoeken (2.630 patiënten) was 93,7 voorvallen per 100 patiëntjaren.
In placebogecontroleerde klinische onderzoeken met achtergrond-DMARD's, was de frequentie van ernstige infecties over een periode van 12/14 weken in de groep die 15 mg upadacitinib kreeg 1,2% vergeleken met 0,6% in de placebogroep. In MTX-gecontroleerde klinische onderzoeken was de frequentie van ernstige infecties over een periode van 12/14 weken in de groep die monotherapie met 15 mg upadacitinib kreeg 0,6% vergeleken met 0,4% in de MTX-groep. De algehele ratio ernstige infecties op lange termijn voor de groep die 15 mg upadacitinib kreeg over alle vijf klinische fase 3-onderzoeken was 3,8 voorvallen per 100 patiëntjaren. De vaakst gemelde ernstige infectie was pneumonie. Het percentage ernstige infecties bleef stabiel bij langdurige blootstelling.
Opportunistische infecties (met uitzondering van tuberculose)
In placebogecontroleerde klinische onderzoeken met achtergrond-DMARD's, was de frequentie van opportunistische infecties over een periode van 12/14 weken in de groep die 15 mg upadacitinib kreeg 0,5% vergeleken met 0,3% in de placebogroep. In MTX-gecontroleerde klinische onderzoeken waren er geen gevallen van opportunistische infecties over een periode van 12/14 weken in de groep die monotherapie met 15 mg upadacitinib kreeg en 0,2% in de MTX-groep. De algehele ratio opportunistische infecties op lange termijn voor de groep die 15 mg upadacitinib kreeg over alle vijf klinische fase 3-onderzoeken was 0,6 voorvallen per 100 patiëntjaren.
De ratio herpes zoster op lange termijn voor de groep die 15 mg upadacitinib kreeg over alle vijf klinische fase 3-onderzoeken was 3,7 voorvallen per 100 patiëntjaren. De meeste gevallen van herpes zoster betroffen één dermatoom en waren niet ernstig van aard.
Verhoogde levertransaminase
In placebogecontroleerde onderzoeken met achtergrond-DMARD's werd tot 12/14 weken in ten minste één meting een verhoging van alanineaminotransferase (ALAT) en aspartaataminotransferase (ASAT) ≥ 3 x bovengrens van normaal (ULN) waargenomen bij respectievelijk 2,1% en 1,5% van de patiënten die werden behandeld met 15 mg upadacitinib, vergeleken met respectievelijk 1,5% en 0,7% van de patiënten die met placebo werden behandeld. Van de 22 gevallen van verhoogde levertransaminase waren de meeste asymptomatisch en van voorbijgaande aard.
In MTX-gecontroleerde onderzoeken werd tot 12/14 weken in ten minste één meting een verhoging van ALAT en ASAT ≥ 3 x ULN waargenomen bij respectievelijk 0,8% en 0,4% van de patiënten die werden behandeld met 15 mg upadacitinib, vergeleken met respectievelijk 1,9% en 0,9% van de patiënten die met MTX werden behandeld.
Het patroon en de incidentie van de verhoging van ALAT/ASAT bleef lange tijd stabiel, met inbegrip van langetermijn-verlengingsonderzoeken.
Verhoogde lipiden
De behandeling met 15 mg upadacitinib werd in verband gebracht met verhogingen in lipideparameters, zoals totaal cholesterol, triglyceriden, LDL-cholesterol en HDL-cholesterol. Er was geen verandering in de LDL/HDL-ratio. De verhoogde waarden werden na 2 tot 4 weken behandeling waargenomen en bleven stabiel bij een langere behandeling. Onder de patiënten in de gecontroleerde onderzoeken met baselinewaarden onder de gespecificeerde limieten werd met de volgende frequenties een verschuiving waargenomen naar boven de gespecificeerde limieten bij ten minste één gelegenheid in 12/14 weken (inclusief patiënten met een geïsoleerde verhoogde waarde):
- Totaal cholesterol ≥ 5,17 mmol/l (200 mg/dl): 62% vs. 31%, in respectievelijk de groep die upadacitinib 15 mg kreeg en de placebogroep
- LDL-cholesterol ≥ 3,36 mmol/l (130 mg/dl): 42% vs. 19%, in respectievelijk de groep die upadacitinib 15 mg kreeg en de placebogroep
- HDL-cholesterol ≥ 1,03 mmol/l (40 mg/dl): 89% vs. 61%, in respectievelijk de groep die upadacitinib 15 mg kreeg en de placebogroep
- Triglyceriden ≥ 2,26 mmol/l (200 mg/dl): 25% vs. 15%, in respectievelijk de groep die upadacitinib 15 mg kreeg en de placebogroep
Creatinekinase (CK-waarden)
In placebogecontroleerde onderzoeken met achtergrond-DMARD's werden gedurende 12/14 weken toenames in CK-waarden waargenomen. Verhoogde CK-waarden > 5 x bovengrens van normaal (ULN) werden over een periode van 12/14 weken gemeld bij 1,0% en 0,3% van de patiënten in respectievelijk de groep die 15 mg upadacitinib kreeg en de placebogroep. De meeste verhoogde waarden van > 5 x ULN waren van voorbijgaande aard en de behandeling hoefde niet te worden gestopt. De gemiddelde CK-waarden waren na 4 weken verhoogd met een gemiddelde toename van 60 U/l na 12 weken en bleven daarna stabiel op de verhoogde waarde, ook gedurende een voortgezette behandeling.
Neutropenie
In placebogecontroleerde onderzoeken met achtergrond-DMARD's werd gedurende 12/14 weken in ten minste één meting een afname in het aantal neutrofielen waargenomen tot onder 1 x 109 cellen/l bij 1,1% en < 0,1% van de patiënten in respectievelijk de groep die 15 mg upadacitinib kreeg en de placebogroep. In klinische onderzoeken werd de behandeling onderbroken als respons op ANC < 1 x 109 cellen/l (zie rubriek 4.2). Het gemiddelde aantal neutrofielen nam over 4 tot 8 weken af. De afname in het aantal neutrofielen bleef op den duur op een lagere waarde dan bij baseline stabiel, ook gedurende een voortgezette behandeling.
Artritis psoriatica
Over het geheel genomen kwam het veiligheidsprofiel bij patiënten met artritis psoriatica die werden behandeld met 15 mg upadacitinib, overeen met het veiligheidsprofiel bij patiënten met reumatoïde artritis. Er werd een hoger percentage ernstige infecties (respectievelijk 2,6 voorvallen per 100 patiëntjaren en 1,3 voorvallen per 100 patiëntjaren) en verhoogde levertransaminasen (ALAT‑verhogingen graad 3 en hoger, respectievelijk 1,4% en 0,4%) waargenomen bij patiënten die behandeld werden met upadacitinib in combinatie met MTX, vergeleken met patiënten die werden behandeld met monotherapie.
Axiale spondyloartritis
Over het geheel genomen kwam het veiligheidsprofiel bij patiënten met axiale spondyloartritis die werden behandeld met 15 mg upadacitinib, overeen met het veiligheidsprofiel bij patiënten met reumatoïde artritis. Er werden geen nieuwe veiligheidsbevindingen vastgesteld.
Reuscelarteriitis
Het veiligheidsprofiel dat werd waargenomen bij patiënten met reuscelarteriitis die werden behandeld met 15 mg upadacitinib, kwam in het algemeen overeen met het veiligheidsprofiel dat bekend is voor upadacitinib.
Ernstige infecties
In het placebogecontroleerde klinische onderzoek was de frequentie van ernstige infecties over een periode van 52 weken in de groep die 15 mg upadacitinib kreeg 5,7% en in de placebogroep 10,7%. Het aantal voorvallen van ernstige infecties op de lange termijn was 5,9 per 100 patiëntjaren bij de 15 mg upadacitinibgroep en 10,5 per 100 patiëntjaren voor de placebogroep.
Opportunistische infecties (met uitzondering van tuberculose)
In het placebogecontroleerde klinische onderzoek was de frequentie van opportunistische infecties (met uitzondering van tuberculose en herpes zoster) over 52 weken in de groep die 15 mg upadacitinib kreeg, 1,9% en in de placebogroep 0,9%. Het aantal langetermijnvoorvallen per 100 patiëntjaren van opportunistische infecties (met uitzondering van tuberculose en herpes zoster) was 1,8 voor de groep die 15 mg upadacitinib kreeg, en 1,5 voor de placebogroep.
In het placebogecontroleerde klinische onderzoek was de frequentie van herpes zoster over 52 weken in de groep die 15 mg upadacitinib kreeg, 5,3% en in de placebogroep 2,7%. Het aantal langetermijnvoorvallen per 100 patiëntjaren van herpes zoster was 5,9 voor de groep die 15 mg upadacitinib kreeg, en 3,0 voor de placebogroep.
Atopische dermatitis
Infecties
In de placebogecontroleerde periode van de klinische onderzoeken was de frequentie van infecties over een periode van 16 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 39% en 43% vergeleken met 30% in de placebogroep. De ratio infecties op lange termijn voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 98,5 en 109,6 voorvallen per 100 patiëntjaren.
In de placebogecontroleerde klinische onderzoeken was de frequentie van ernstige infecties over een periode van 16 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 0,8% en 0,4% vergeleken met 0,6% in de placebogroep. De ratio ernstige infecties op lange termijn voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 2,3 en 2,8 voorvallen per 100 patiëntjaren.
Opportunistische infecties (met uitzondering van tuberculose)
In de placebogecontroleerde periode van de klinische onderzoeken waren alle gemelde opportunistische infecties (exclusief tb en herpes zoster) eczema herpeticum. De frequentie van eczema herpeticum over een periode van 16 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 0,7% en 0,8% vergeleken met 0,4% in de placebogroep. De ratio eczema herpeticum op lange termijn voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 1,6 en 1,8 voorvallen per 100 patiëntjaren. Er is één geval van oesofageale candidiasis gemeld met 30 mg upadacitinib.
De ratio herpes zoster op lange termijn voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 3,5 en 5,2 voorvallen per 100 patiëntjaren. De meeste gevallen van herpes zoster betroffen één dermatoom en waren niet ernstig van aard.
Afwijkende laboratoriumwaarden
Dosisafhankelijke veranderingen in verhoogd ALAT en/of verhoogd ASAT (≥ 3 x ULN), lipideparameters, CK-waarden, (> 5 x ULN) en neutropenie (ANC < 1 x 109 cellen/l die samenhangen met behandeling met upadacitinib waren vergelijkbaar met wat werd waargenomen in de klinische onderzoeken naar reumatologische ziekte.
Kleine verhogingen in LDL-cholesterol werden na week 16 waargenomen in onderzoeken naar atopische dermatitis. In week 52 was de gemiddelde stijging van het LDL-cholesterol ten opzichte van baseline 0,41 mmol/l voor 15 mg upadacitinib en 0,56 mmol/l voor 30 mg upadacitinib.
Alopecia areata
Het veiligheidsprofiel van upadacitinib 15 mg en 30 mg dat is waargenomen bij patiënten met alopecia areata, kwam in het algemeen overeen met het bekende veiligheidsprofiel bij patiënten met atopische dermatitis. Er werden geen nieuwe veiligheidsbevindingen vastgesteld.
Vitiligo
Het veiligheidsprofiel van upadacitinib 15 mg dat is waargenomen bij patiënten met vitiligo, kwam in het algemeen overeen met het bekende veiligheidsprofiel bij patiënten met atopische dermatitis. Er werden geen nieuwe veiligheidsbevindingen vastgesteld. Er werd een hogere incidentie van hypercholesterolemie gezien bij patiënten met vitiligo die werden behandeld met upadacitinib 15 mg (3,4%) vergeleken met placebo (2,0%).
Colitis ulcerosa
Het algehele veiligheidsprofiel dat werd waargenomen bij patiënten met colitis ulcerosa, kwam in het algemeen overeen met het veiligheidsprofiel dat werd waargenomen bij patiënten met reumatoïde artritis.
Bij een inductieperiode van 16 weken werd een hoger percentage herpes zoster waargenomen dan bij een inductieperiode van 8 weken.
Infecties
In de placebogecontroleerde inductieonderzoeken was de frequentie van infecties over een periode van 8 weken in de groep die 45 mg upadacitinib kreeg 20,7% en in de placebogroep 17,5%. In het placebogecontroleerde onderhoudsonderzoek was de frequentie van infecties over een periode van 52 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 40,4% en 44,2%, en in de placebogroep 38,8%. Het aantal voorvallen van infecties op de langere termijn bij 15 mg en 30 mg upadacitinib was respectievelijk 64,5 en 77,8 per 100 patiëntjaren.
In de placebogecontroleerde inductieonderzoeken was de frequentie van ernstige infecties over een periode van 8 weken in zowel de groep die 45 mg upadacitinib kreeg als de placebogroep 1,3%. Er werden geen additionele ernstige infecties waargenomen tijdens een verlengde behandeling van 8 weken met 45 mg upadacitinib. In het placebogecontroleerde onderhoudsonderzoek was de frequentie van ernstige infecties over een periode van 52 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 3,6% en 3,2%, ten opzichte van 3,3% in de placebogroep. Het aantal langetermijnvoorvallen per 100 patiëntjaren van ernstige infecties voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 3,0 en 4,6. In de inductie- en onderhoudsfase was de ernstige infectie die het vaakst werd gemeld, pneumonie door COVID‑19.
Opportunistische infecties (met uitzondering van tuberculose)
In de placebogecontroleerde inductieonderzoeken was de frequentie van opportunistische infecties (met uitzondering van tuberculose en herpes zoster) over een periode van 8 weken in de groep die 45 mg upadacitinib kreeg, 0,4% en in de placebogroep 0,3%. Er werden geen extra opportunistische infecties (met uitzondering van tuberculose en herpes zoster) waargenomen tijdens de verlengde behandeling van 8 weken met 45 mg upadacitinib. In het placebogecontroleerde onderhoudsonderzoek was de frequentie van opportunistische infecties (met uitzondering van tuberculose en herpes zoster) over een periode van 52 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 0,8% en 0,8% ten opzichte van 0,8% in de placebogroep. Het aantal langetermijnvoorvallen per 100 patiëntjaren van opportunistische infecties (met uitzondering van tuberculose en herpes zoster) voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 0,3 en 0,6.
In de placebogecontroleerde inductieonderzoeken over een periode van 8 weken was de frequentie van herpes zoster in de groep die 45 mg upadacitinib kreeg 0,6% en in de placebogroep 0%. De frequentie van herpes zoster tijdens een 16 weken durende behandeling met 45 mg upadacitinib was 3,9%. In het placebogecontroleerde onderhoudsonderzoek was de frequentie van herpes zoster over een periode van 52 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 4,8% en 5,6% ten opzichte van 0% in de placebogroep. Het aantal langetermijnvoorvallen per 100 patiëntjaren van herpes zoster voor de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 4,5 en 7,2.
Gastro-intestinale perforaties
In de placebogecontroleerde onderhoudsperiode werd gastro-intestinale perforatie gemeld bij 1 patiënt behandeld met placebo (1,5 per 100 patiëntjaren) en bij geen enkele patiënt behandeld met upadacitinib 15 mg of 30 mg. In het langetermijn-verlengingsonderzoek rapporteerden 1 patiënt behandeld met upadacitinib 15 mg (0,1 per 100 patiëntjaren) en 1 patiënt behandeld met upadacitinib 30 mg (<0,1 per 100 patiëntjaren) voorvallen.
Afwijkende laboratoriumwaarden
In de klinische onderzoeken naar de inductiedosis en onderhoudsdosis waren de veranderingen in de laboratoriumwaarden voor verhoogde ALAT en/of verhoogde ASAT (≥ 3 x ULN), CK-waarden (> 5 x ULN) en neutropenie (ANC < 1 x 109 cellen/l) die werden geassocieerd met de behandeling met upadacitinib, in het algemeen vergelijkbaar met wat werd waargenomen in de klinische onderzoeken naar reumatologische aandoeningen en atopische dermatitis. Er werden dosisafhankelijke veranderingen in deze laboratoriumparameters waargenomen die werden geassocieerd met een behandeling van 15 mg en 30 mg upadacitinib.
In de placebogecontroleerde inductieonderzoeken die maximaal 8 weken duurden, was bij 2,0% van de patiënten die 45 mg upadacitinib kregen en bij 0,8% van de patiënten die een placebo kregen, bij minimaal één meting het aantal lymfocyten tot onder 0,5 x 109 cellen/l gedaald. In het placebogecontroleerde onderhoudsonderzoek dat maximaal 52 weken duurde, was bij 1,6% van de patiënten die 15 mg upadacitinib kregen, bij 1,2% van de patiënten die 30 mg upadacitinib kregen en bij 0,8% van de patiënten die een placebo kregen, bij minimaal één meting het aantal lymfocyten tot onder 0,5 x 109 cellen/l gedaald. In de klinische onderzoeken werd de behandeling onderbroken als ALC < 0,5 x 109 cellen/l (zie rubriek 4.2). Er werden geen opmerkelijke gemiddelde veranderingen in het aantal lymfocyten waargenomen gedurende de behandeling met upadacitinib.
Er werden verhogingen van lipidenparameters waargenomen bij een behandeling van 8 weken met 45 mg upadacitinib en deze bleven in het algemeen stabiel bij een langere behandeling met 15 mg of 30 mg upadacitinib. Onder de patiënten in de placebogecontroleerde inductieonderzoeken met baselinewaarden onder de gespecificeerde limieten, werd met de volgende frequenties een verschuiving waargenomen naar een waarde boven de gespecificeerde limieten bij ten minste één gelegenheid in 8 weken (inclusief patiënten met een geïsoleerde verhoogde waarde):
- Totaal cholesterol ≥ 5,17 mmol/l (200 mg/dl): 49% versus 11% in respectievelijk de groep die 45 mg upadacitinib kreeg en de placebogroep
- LDL-cholesterol ≥ 3,36 mmol/l (130 mg/dl): 27% versus 9% in respectievelijk de groep die 45 mg upadacitinib kreeg en de placebogroep
- HDL-cholesterol ≥ 1,03 mmol/l (40 mg/dl): 79% versus 36% in respectievelijk de groep die 45 mg upadacitinib kreeg en de placebogroep
- Triglyceriden ≥ 2,26 mmol/l (200 mg/dl): 6% versus 4% in respectievelijk de groep die 45 mg upadacitinib kreeg en de placebogroep
Ziekte van Crohn
Het veiligheidsprofiel dat werd waargenomen bij met upadacitinib behandelde patiënten met de ziekte van Crohn, kwam in het algemeen overeen met het bekende veiligheidsprofiel voor upadacitinib.
Ernstige infecties
In de placebogecontroleerde inductieonderzoeken was de frequentie van ernstige infectie over een periode van 12 weken in de groep die 45 mg upadacitinib kreeg 1,9% en in de placebogroep 1,7%. In het placebogecontroleerde onderhoudsonderzoek was de frequentie van ernstige infectie over een periode van 52 weken in de groepen die 15 mg en 30 mg upadacitinib kregen, respectievelijk 3,2% en 5,7%, vergeleken met 4,5% in de placebogroep. Het aantal voorvallen van ernstige infecties op de langere termijn in de groepen die 15 mg en 30 mg upadacitinib kregen, was respectievelijk 5,1 en 7,3 per 100 patiëntjaren bij patiënten die reageerden op 45 mg upadacitinib als inductiebehandeling. De vaakst gemelde ernstige infecties in de inductie‑ en onderhoudsonderzoeken waren gastro‑intestinale infecties.
Gastro-intestinale perforaties
Tijdens de placebogecontroleerde periode in de klinische fase 3 inductie-onderzoeken werd gedurende 12 weken bij 1 patiënt (0,1%) die werd behandeld met 45 mg upadacitinib gastro-intestinale perforatie gemeld en bij geen enkele patiënt met placebo. Bij alle patiënten die tijdens de inductieonderzoeken werden behandeld met 45 mg upadacitinib (n=938), werd gastro-intestinale perforatie gemeld bij 4 patiënten (0,4%).
In de placebogecontroleerde langetermijnperiode werd gastro-intestinale perforatie gemeld bij 1 patiënt die werd behandeld met placebo (0,7 per 100 patiëntjaren), bij 1 patiënt die werd behandeld met 15 mg upadacitinib (0,4 per 100 patiëntjaren) en bij 1 patiënt die werd behandeld met 30 mg upadacitinib (0,4 per 100 patiëntjaren). Bij alle patiënten die werden behandeld met 30 mg upadacitinib als rescuebehandeling (n=336) werd gastro-intestinale perforatie gemeld bij 3 patiënten (0,8 per 100 patiëntjaren) tijdens langdurige behandeling.
Afwijkende laboratoriumwaarden
In de klinische onderzoeken naar de inductiedosis en onderhoudsdosis waren de veranderingen in de laboratoriumwaarden voor verhoogde ALAT en/of verhoogde ASAT (≥ 3 x ULN), CK‑waarden (> 5 x ULN), neutropenie (ANC < 1 x 109 cellen/l) en lipidenparameters die werden geassocieerd met de behandeling met upadacitinib, in het algemeen vergelijkbaar met wat werd waargenomen in de klinische onderzoeken naar reumatologische aandoeningen, atopische dermatitis en colitis ulcerosa. Er werden dosisafhankelijke veranderingen in deze laboratoriumparameters waargenomen die werden geassocieerd met een behandeling van 15 mg en 30 mg upadacitinib.
In de placebogecontroleerde inductieonderzoeken die maximaal 12 weken duurden, was bij 2,2% van de patiënten die 45 mg upadacitinib kregen en bij 2,0% van de patiënten die een placebo kregen, bij minimaal één meting het aantal lymfocyten tot onder 0,5 x 109 cellen/l gedaald. In het placebogecontroleerde onderhoudsonderzoek dat maximaal 52 weken duurde, was bij 4,6% van de patiënten die 15 mg upadacitinib kregen, bij 5,2% van de patiënten die 30 mg upadacitinib kregen en bij 1,8% van de patiënten die een placebo kregen, bij minimaal één meting het aantal lymfocyten tot onder 0,5 x 109 cellen/l gedaald. In de klinische onderzoeken werd de behandeling onderbroken als ALC < 0,5 x 109 cellen/l (zie rubriek 4.2). Er werden geen opmerkelijke gemiddelde veranderingen in het aantal lymfocyten waargenomen gedurende de behandeling met upadacitinib in de loop van de tijd.
In de placebogecontroleerde inductieonderzoeken die maximaal 12 weken duurden, was bij 2,7% van de patiënten in de groep die 45 mg upadacitinib kreeg en bij 1,4% van de patiënten in de groep die een placebo kreeg de hemoglobineconcentratie bij minimaal één meting tot onder 8 g/dl gedaald. In het placebogecontroleerde onderhoudsonderzoek dat maximaal 52 weken duurde, was bij 1,4% van de patiënten in de groep die 15 mg upadacitinib kreeg, bij 4,4% van de patiënten in de groep die 30 mg upadacitinib kreeg en bij 2,8% van de patiënten in de groep die een placebo kreeg de hemoglobineconcentratie bij minimaal één meting tot onder 8 g/dl gedaald. In de klinische onderzoeken werd de behandeling onderbroken als Hb < 8 g/dl (zie rubriek 4.2). Er werden geen opmerkelijke gemiddelde veranderingen in de hemoglobineconcentratie waargenomen gedurende de behandeling met upadacitinib.
Ouderen
Op basis van beperkte gegevens bij patiënten van 65 jaar en ouder met atopische dermatitis, colitis ulcerosa en de ziekte van Crohn kwamen bijwerkingen in het algemeen vaker voor bij een dosis van 30 mg upadacitinib dan bij een dosis van 15 mg upadacitinib (zie rubriek 4.4).
Er waren geen gegevens beschikbaar van patiënten vanaf 65 jaar met ernstige alopecia areata.
Van de 612 patiënten die werden behandeld in de klinische fase 3-onderzoeken naar vitiligo waren er 55 in de leeftijd van 65 jaar of ouder. 36 van deze patiënten werden behandeld met upadacitinib 15 mg tijdens de placebogecontroleerde periode. In het algemeen waren de percentages ernstige en hevige bijwerkingen bij patiënten van 65 jaar of ouder vergelijkbaar in de groep met 15 mg upadacitinib en de groep met placebo.
Pediatrische patiënten
Atopische dermatitis
In totaal werden 541 adolescenten van 12 tot en met 17 jaar met atopische dermatitis behandeld in de wereldwijde fase 3‑onderzoeken (n=343) en de aanvullende adolescentensubstudies (n=198). Van hen werden 264 adolescenten blootgesteld aan 15 mg en 265 aan 30 mg upadacitinib. Het veiligheidsprofiel van 15 mg en 30 mg upadacitinib bij adolescenten was vergelijkbaar met dat bij volwassenen. Bij langdurige blootstelling werd de bijwerking huidpapilloom gerapporteerd bij respectievelijk 3,4% en 6,8% van de adolescenten met atopische dermatitis in de upadacitinib 15 mg en 30 mg-groepen.
Alopecia areata
In totaal werden 117 adolescenten van 12 tot en met 17 jaar met een gewicht van minstens 30 kg met alopecia areata behandeld met upadacitinib in de placebogecontroleerde fase 3-onderzoeken. Van hen werden er 62 blootgesteld aan 15 mg en 57 aan 30 mg. Het veiligheidsprofiel van upadacitinib 15 mg en 30 mg bij adolescenten was vergelijkbaar met dat bij volwassenen.
Vitiligo
In totaal werden 52 adolescenten met vitiligo, in de leeftijd van 12 tot en met 17 jaar en een gewicht van minstens 30 kg, in de fase 3-onderzoeken behandeld met upadacitinib 15 mg. 37 van deze adolescenten kregen upadacitinib 15 mg tijdens de placebogecontroleerde periode. Het veiligheidsprofiel van upadacitinib 15 mg dat werd gezien bij adolescente patiënten met vitiligo was in het algemeen vergelijkbaar met het bekende veiligheidsprofiel bij adolescente patiënten met atopische dermatitis. Er werden geen nieuwe veiligheidsbevindingen vastgesteld. Er werden geen ernstige bijwerkingen gemeld bij de adolescente populatie die werd behandeld met upadacitinib tijdens de placebogecontroleerde periode.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
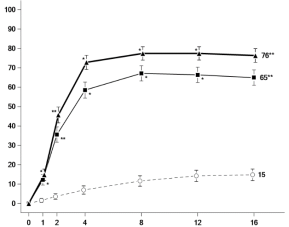
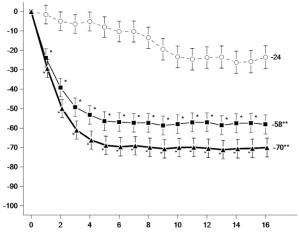
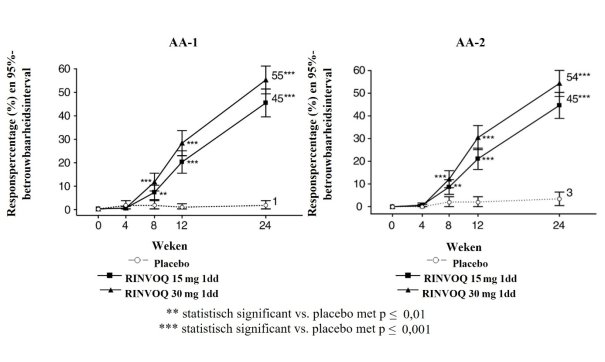
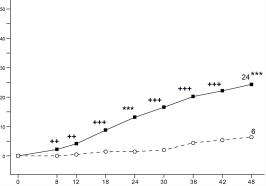
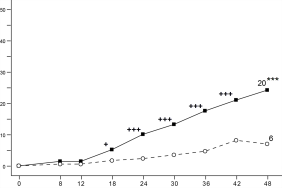
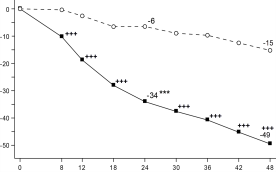
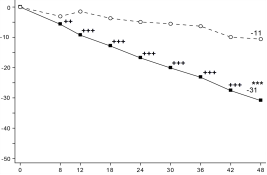
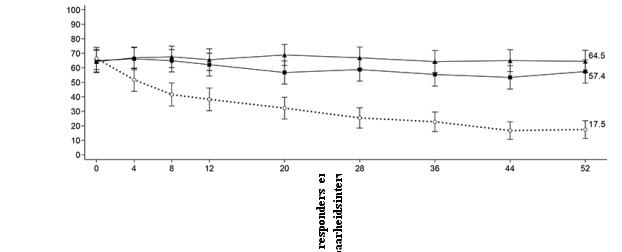
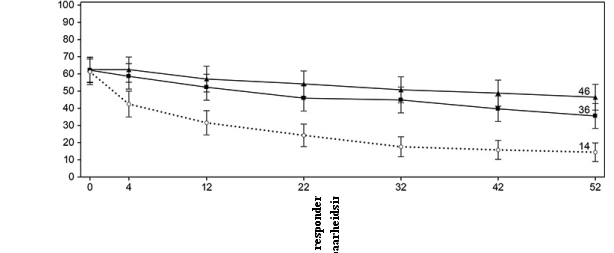 Figuur 6 Aandeel patiënten dat klinische remissie bereikt in onderhoudsonderzoek CD‑3
Figuur 6 Aandeel patiënten dat klinische remissie bereikt in onderhoudsonderzoek CD‑37. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
AbbVie Deutschland GmbH & Co. KG
Knollstrasse
67061 Ludwigshafen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/19/1404/001
EU/1/19/1404/002
EU/1/19/1404/003
EU/1/19/1404/004
EU/1/19/1404/005
EU/1/19/1404/006
EU/1/19/1404/007
EU/1/19/1404/008
EU/1/19/1404/009
EU/1/19/1404/010
EU/1/19/1404/011
10. DATUM VAN HERZIENING VAN DE TEKST
07/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3963147 | RINVOQ 15MG VERLENGDE AFGIFTE TABL 28 | L04AA44 | € 822,46 | - | Ja | € 12,8 | € 8,5 |
| 3963154 | RINVOQ 15MG VERLENGDE AFGIFTE TABL 98 | L04AA44 | € 2505,77 | - | Ja | € 15,9 | € 10,5 |
| 4404877 | RINVOQ 30MG VERLENGDE AFGIFTE TABL 28 | L04AA44 | € 1221,79 | - | Ja | € 12,8 | € 8,5 |
| 4404885 | RINVOQ 30MG VERLENGDE AFGIFTE TABL 98 | L04AA44 | € 3401,28 | - | Ja | € 15,9 | € 10,5 |
| 4550802 | RINVOQ 45MG VERLENGDE AFGIFTE TABL 28 | € 1852,75 | - | Ja | € 12,8 | € 8,5 |