1. NAAM VAN HET GENEESMIDDEL
Tremfya 100 mg OnePress oplossing voor injectie in een voorgevulde pen
Tremfya 100 mg PushPen oplossing voor injectie in een voorgevulde pen
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Tremfya 100 mg OnePress oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen bevat 100 mg guselkumab in 1 ml oplossing.
Tremfya 100 mg PushPen oplossing voor injectie in een voorgevulde pen
Elke voorgevulde pen bevat 100 mg guselkumab in 1 ml oplossing.
Guselkumab is een geheel humaan immunoglobuline-G1-lambda (IgG1λ)-monoklonaal antilichaam (mAb) en wordt met behulp van recombinant-DNA-technologie geproduceerd in ovariumcellen van de Chinese hamster (CHO-cellen).
Hulpstof(fen) met bekend effect
Dit geneesmiddel bevat 0,5 mg polysorbaat 80 (E433) in elke voorgevulde pen. Dit komt overeen met 0,5 mg/ml.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie (injectie)
De oplossing is helder en kleurloos tot lichtgeel en kan enkele kleine witte of doorzichtige deeltjes bevatten, met een streef-pH van 5,8 en een osmolariteit van ongeveer 367,5 mOsm/l.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Plaque psoriasis bij volwassenen
Tremfya is geïndiceerd voor de behandeling van matige tot ernstige plaque psoriasis bij volwassenen die in aanmerking komen voor systemische therapie.
Arthritis psoriatica
Tremfya, alleen of in combinatie met methotrexaat (MTX), is geïndiceerd voor de behandeling van actieve arthritis psoriatica bij volwassen patiënten die onvoldoende reageren op of intolerant zijn geweest voor een eerdere behandeling met een disease-modifying antirheumatic drug (DMARD) (zie rubriek 5.1).
Colitis ulcerosa
Tremfya is geïndiceerd voor de behandeling van volwassen patiënten met matig tot ernstig actieve colitis ulcerosa die onvoldoende of niet meer reageren op ofwel conventionele therapie ofwel een biologische behandeling of deze behandelingen niet verdragen.
Ziekte van Crohn
Tremfya is geïndiceerd voor de behandeling van volwassen patiënten met matig tot ernstig actieve ziekte van Crohn die onvoldoende of niet meer reageren op ofwel conventionele therapie ofwel een biologische behandeling of deze behandelingen niet verdragen.
4.2 Dosering en wijze van toediening
Dit geneesmiddel is bedoeld voor gebruik onder begeleiding en supervisie van een arts met ervaring in het diagnosticeren en behandelen van aandoeningen waarvoor het geïndiceerd is.
Dosering
Plaque psoriasis
De aanbevolen dosis is 100 mg via subcutane injectie in week 0 en week 4, gevolgd door een onderhoudsdosis eenmaal per 8 weken (q8w).
Bij patiënten bij wie er na 16 weken behandeling geen respons is vastgesteld, dient te worden overwogen om de behandeling te stoppen.
Arthritis psoriatica
De aanbevolen dosis is 100 mg via subcutane injectie in week 0 en week 4, gevolgd door een onderhoudsdosis eenmaal per 8 weken. Voor patiënten die op basis van klinisch oordeel een hoog risico hebben op gewrichtsschade, kan een dosis van 100 mg elke 4 weken (q4w) worden overwogen (zie rubriek 5.1).
Bij patiënten die na 24 weken behandeling geen respons hebben vertoond, dient te worden overwogen om de behandeling te stoppen.
Colitis ulcerosa
Een van de volgende twee inductiedoseringsschema’s wordt aanbevolen:
- 200 mg toegediend via intraveneuze infusie in week 0, week 4 en week 8. Raadpleeg de SmPC voor Tremfya 200 mg concentraat voor oplossing voor infusie.
of
- 400 mg toegediend via subcutane injectie (gegeven als twee opeenvolgende injecties van elk 200 mg) in week 0, week 4 en week 8. Raadpleeg de SmPC voor Tremfya 200 mg oplossing voor injectie.
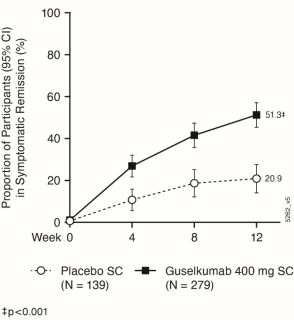
Na voltooiing van het inductiedoseringsschema is de aanbevolen onderhoudsdosis vanaf week 16 100 mg toegediend via subcutane injectie elke 8 weken (q8w). Als alternatief kan voor patiënten die volgens klinisch oordeel onvoldoende therapeutisch voordeel hebben bij de inductiebehandeling een onderhoudsdosis van 200 mg worden overwogen, toegediend via subcutane injectie vanaf week 12 en vervolgens elke 4 weken (q4w) (zie rubriek 5.1). Raadpleeg voor de dosis van 200 mg de SmPC voor Tremfya 200 mg oplossing voor injectie.
Immunomodulatoren en/of corticosteroïden mogen worden voortgezet tijdens de behandeling met guselkumab. Bij patiënten die respons hebben vertoond op behandeling met guselkumab, kunnen corticosteroïden worden verminderd of stopgezet volgens de zorgstandaard.
Er dient overwogen te worden om de behandeling stop te zetten bij patiënten bij wie na 24 weken behandeling geen therapeutisch voordeel is aangetoond.
Ziekte van Crohn
Een van de volgende twee inductiedoseringsschema’s wordt aanbevolen:
- 200 mg toegediend via intraveneuze infusie in week 0, week 4 en week 8. Raadpleeg de SmPC voor Tremfya 200 mg concentraat voor oplossing voor infusie.
of
- 400 mg toegediend via subcutane injectie (gegeven als twee opeenvolgende injecties van elk 200 mg) in week 0, week 4 en week 8. Raadpleeg de SmPC voor Tremfya 200 mg oplossing voor injectie.
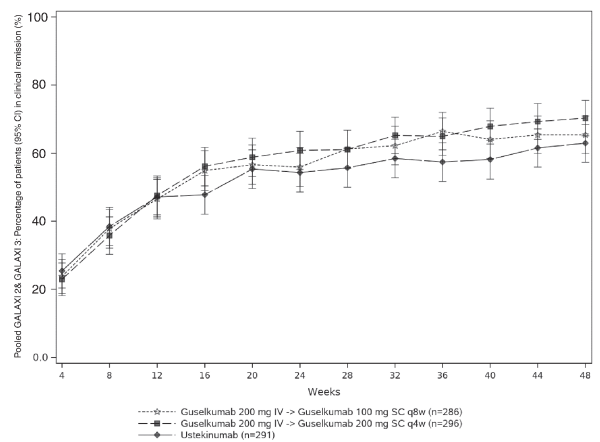
Na voltooiing van het inductiedoseringsschema is de aanbevolen onderhoudsdosis vanaf week 16 100 mg toegediend via subcutane injectie elke 8 weken (q8w). Als alternatief kan voor patiënten die volgens klinisch oordeel onvoldoende therapeutisch voordeel hebben bij de inductiebehandeling een onderhoudsdoseringsschema van 200 mg worden overwogen, toegediend via subcutane injectie vanaf week 12 en vervolgens elke 4 weken (q4w) (zie rubriek 5.1). Raadpleeg voor de dosis van 200 mg de SmPC voor Tremfya 200 mg oplossing voor injectie.
Immunomodulatoren en/of corticosteroïden mogen worden voortgezet tijdens de behandeling met guselkumab. Bij patiënten die respons hebben vertoond op behandeling met guselkumab, kunnen corticosteroïden worden verminderd of stopgezet volgens de zorgstandaard.
Er dient overwogen te worden om de behandeling stop te zetten bij patiënten bij wie na 24 weken behandeling geen therapeutisch voordeel is aangetoond.
Gemiste dosis
Als een dosis wordt overgeslagen, moet de dosis zo snel mogelijk worden toegediend. Daarna moet de toediening op het normale geplande tijdstip worden hervat.
Bijzondere populaties
Ouderen
De dosis hoeft niet te worden aangepast (zie rubriek 5.2).
Er is beperkte informatie bij patiënten met een leeftijd van ≥ 65 jaar en zeer beperkte informatie bij patiënten met een leeftijd van ≥ 75 jaar (zie rubriek 5.2).
Nier- of leverinsufficiëntie
Tremfya is niet bij deze patiëntengroepen onderzocht. Over het algemeen wordt niet verwacht dat deze aandoeningen significante invloed zullen hebben op de farmacokinetiek van monoklonale antilichamen, en dosisaanpassingen worden niet noodzakelijk geacht. Zie rubriek 5.2 voor verdere informatie over de eliminatie van guselkumab.
Pediatrische patiënten
De veiligheid en werkzaamheid van Tremfya zijn niet vastgesteld bij patiënten jonger dan 18 jaar met colitis ulcerosa, de ziekte van Crohn en arthritis psoriatica en bij patiënten jonger dan 6 jaar met psoriasis. Er zijn geen gegevens beschikbaar. Tremfya 100 mg oplossing voor injectie in voorgevulde pennen wordt niet aanbevolen voor gebruik bij kinderen jonger dan 18 jaar vanwege onvoldoende gegevens over de veiligheid en werkzaamheid. De momenteel beschikbare gegevens met andere presentaties worden beschreven in rubrieken 4.8, 5.1 en 5.2.
Wijze van toediening
Uitsluitend subcutaan gebruik. De injectieplaatsen zijn onder andere de buik, de dij en de achterkant van de bovenarm. Tremfya mag niet worden geïnjecteerd op plaatsen waar de huid gevoelig, gekneusd, rood, hard, dik of schilferig is. Door psoriasis aangetaste huid dient zo mogelijk te worden vermeden als injectieplaats.
Na een adequate training in de techniek van het subcutaan injecteren mogen patiënten Tremfya injecteren als een arts beslist dat dit aangewezen is. De arts dient echter te zorgen voor een adequate medische opvolging van de patiënten.
Patiënten dienen geïnstrueerd te worden de volledige hoeveelheid oplossing te injecteren, overeenkomstig de in de doos bijgesloten 'Instructies voor gebruik'.
Voor instructies over bereiding van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra-indicaties
Ernstige overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Klinisch relevante actieve infecties (bijv. actieve tuberculose, zie rubriek 4.4).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest voorkomende bijwerking was infecties van de luchtwegen (ongeveer 8% van de patiënten in studies bij colitis ulcerosa, 11% van de patiënten in studies bij de ziekte van Crohn en 15% van de patiënten in de klinische studies bij psoriasis en bij arthritis psoriatica).
Het algemene veiligheidsprofiel bij patiënten die worden behandeld met Tremfya is vergelijkbaar voor patiënten met psoriasis, arthritis psoriatica, colitis ulcerosa en de ziekte van Crohn.
Bijwerkingen in tabelvorm
Tabel 1 toont een lijst van bijwerkingen uit klinische studies bij psoriasis, arthritis psoriatica, colitis ulcerosa en de ziekte van Crohn en bijwerkingen gemeld uit post-marketingervaring. De bijwerkingen zijn ingedeeld volgens de MedDRA systeem/orgaanklassen en frequenties, met de volgende definities: zeer vaak (≥1/10); vaak (≥1/100, <1/10); soms (≥1/1.000, <1/100); zelden (≥1/10.000, <1/1.000); zeer zelden (<1/10.000), niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiegroep zijn de bijwerkingen gerangschikt in volgorde van afnemende ernst.
Tabel 1: Lijst van bijwerkingen | ||
Systeem/orgaanklasse | Frequentie | Bijwerkingen |
Infecties en parasitaire aandoeningen | Zeer vaak | Luchtweginfecties |
Soms | Herpes simplex infecties | |
Soms | Tinea-infecties | |
Soms | Gastro-enteritis | |
Immuunsysteemaandoeningen | Zelden | Overgevoeligheid |
Zelden | Anafylaxie | |
Zenuwstelselaandoeningen | Vaak | Hoofdpijn |
Maagdarmstelselaandoeningen | Vaak | Diarree |
Huid- en onderhuidaandoeningen | Vaak | Rash |
Soms | Urticaria | |
Skeletspierstelsel- en bindweefselaandoeningen | Vaak | Artralgie |
Algemene aandoeningen en toedieningsplaatsstoornissen | Vaak | Injectieplaatsreacties |
Onderzoeken | Vaak | Transaminasen verhoogd |
Soms | Neutrofielentelling verlaagd | |
Beschrijving van geselecteerde bijwerkingen
Transaminasen verhoogd
In twee klinische fase III-studies bij arthritis psoriatica werd in de placebogecontroleerde periode de bijwerking transaminasen verhoogd (dit omvat ALAT verhoogd, ASAT verhoogd, leverenzym verhoogd, transaminasen verhoogd, leverfunctietest abnormaal, hypertransaminasemie) vaker gemeld in de met guselkumab behandelde groepen (8,6% in de 100 mg subcutane q4w-groep en 8,3% in de 100 mg subcutane q8w-groep) dan in de placebogroep (4,6%). In de loop van 1 jaar werd de bijwerking transaminasen verhoogd (zoals hierboven) gemeld bij 12,9% van de patiënten in de q4w-groep en bij 11,7% van de patiënten in de q8w-groep.
Op basis van laboratoriummetingen waren de meeste transaminaseverhogingen (ALAT en ASAT) ≤ 3 x de bovengrens van normaal (upper limit of normal; ULN). Transaminaseverhogingen van > 3 tot ≤ 5 x ULN en > 5 x ULN kwamen weinig voor en traden in de guselkumab-q4w-groep vaker op dan in de guselkumab-q8w-groep (tabel 2). Een vergelijkbaar frequentiepatroon naar ernst en naar behandelgroep werd waargenomen tot en met het eind van de 2 jaar durende klinische fase III-studie bij arthritis psoriatica.
Tabel 2: Frequentie van patiënten met post-baseline transaminaseverhogingen in twee klinische fase III‑studies bij arthritis psoriatica | |||||
| T/m week 24a | T/m 1 jaarb | |||
Placebo | guselkumab | guselkumab | guselkumab | guselkumab | |
ALAT | |||||
>1 tot ≤3 x ULN | 30,0% | 28,2% | 35,0% | 33,5% | 41,2% |
>3 tot ≤5 x ULN | 1,4% | 1,1% | 2,7% | 1,6% | 4,6% |
>5 x ULN | 0,8% | 0,8% | 1,1% | 1,1% | 1,1% |
ASAT | |||||
>1 tot ≤3 x ULN | 20,0% | 18,8% | 21,6% | 22,8% | 27,8% |
>3 tot ≤5 x ULN | 0,5% | 1,6% | 1,6% | 2,9% | 3,8% |
>5 x ULN | 1,1% | 0,5% | 1,6% | 0,5% | 1,6% |
a placebogecontroleerde periode. | |||||
De frequentie van transaminaseverhogingen (ALAT en ASAT) voor de guselkumab-q8w-dosis was in de klinische studies bij psoriasis, gedurende 1 jaar, gelijk aan wat werd waargenomen voor de guselkumab-q8w-dosis in klinische studies bij arthritis psoriatica. Gedurende 5 jaar nam de incidentie van transaminaseverhoging per jaar behandeling met guselkumab niet toe. De meeste transaminaseverhogingen waren ≤ 3 x ULN.
In de meeste gevallen was de verhoging in transaminasen voorbijgaand en leidde deze niet tot het stoppen met de behandeling.
In samengevoegde klinische fase II- en III-studies bij de ziekte van Crohn werd in de placebogecontroleerde inductieperiode (week 0-12) de bijwerking transaminasen verhoogd (dit omvat ALAT verhoogd, ASAT verhoogd, leverenzym verhoogd, transaminasen verhoogd en leverfunctietest verhoogd) vaker gemeld in de met guselkumab behandelde groepen (1,7% van de patiënten) dan in de placebogroep (0,6% van de patiënten). In samengevoegde klinische fase II- en III-studies bij de ziekte van Crohn werd in de rapportageperiode van ongeveer een jaar de bijwerking transaminasen verhoogd (dit omvat ALAT verhoogd, ASAT verhoogd, leverenzym verhoogd, transaminasen verhoogd, leverfunctietest abnormaal en leverfunctietest verhoogd) gemeld bij 3,4% van de patiënten in de groep behandeld met guselkumab 200 mg subcutaan q4w en bij 4,1% van de patiënten in de groep behandeld met guselkumab 100 mg subcutaan q8w, tegenover 2,4% in de placebogroep.
Op basis van laboratoriummetingen in samengevoegde klinische fase II- en III-studies bij de ziekte van Crohn was de frequentie van ALAT- of ASAT-verhogingen lager dan die waargenomen in klinische fase III-studies bij arthritis psoriatica. In samengevoegde klinische fase II- en III-studies bij de ziekte van Crohn werden in de placebogecontroleerde periode (week 12) ALAT-verhoging (< 1% van de patiënten) en ASAT-verhoging (< 1% van de patiënten) ≥ 3 x ULN gemeld bij patiënten behandeld met guselkumab. In samengevoegde klinische fase II- en III-studies bij de ziekte van Crohn werden in de rapportageperiode van ongeveer een jaar ALAT-verhoging en/of ASAT-verhoging ≥ 3 x ULN gemeld bij 2,7% van de patiënten in de groep behandeld met guselkumab 200 mg subcutaan q4w en bij 2,6% van de patiënten in de groep behandeld met guselkumab 100 mg subcutaan q8w, tegenover 1,9% in de placebogroep. In de meeste gevallen was de verhoging in transaminasen voorbijgaand en leidde deze niet tot het stoppen met de behandeling.
Neutrofielentelling verlaagd
In twee klinische fase III-studies bij arthritis psoriatica werd in de placebogecontroleerde periode de bijwerking verlaagde neutrofielentelling vaker gemeld in de met guselkumab behandelde groepen (0,9%) dan in de placebogroep (0%). In de loop van 1 jaar werd de bijwerking verlaagde neutrofielentelling gemeld bij 0,9% van de met guselkumab behandelde patiënten. In de meeste gevallen was de afname van de neutrofielentelling in het bloed licht, voorbijgaand, niet geassocieerd met infectie en leidde deze niet tot het stoppen met de behandeling.
Gastro-enteritis
In twee klinische fase III-studies bij psoriasis trad tijdens de placebogecontroleerde periode vaker gastro-enteritis op in de groep die werd behandeld met guselkumab (1,1%) dan in de placebogroep (0,7%). Tot en met week 264 meldde 5,8% van alle met guselkumab behandelde patiënten gastro-enteritis. De bijwerkingen van gastro-enteritis waren niet ernstig en leidden t/m week 264 niet tot stopzetting van guselkumab. De frequenties van gastro-enteritis waargenomen in klinische studies bij arthritis psoriatica in de placebogecontroleerde periode waren gelijk aan die waargenomen in de klinische studies bij psoriasis.
Injectieplaatsreacties
In twee klinische fase III-studies bij psoriasis kwamen t/m week 48 bij 0,7% van de injecties met guselkumab en bij 0,3% van de injecties met placebo injectieplaatsreacties voor. Tot en met week 264 ging 0,4% van de guselkumab-injecties gepaard met injectieplaatsreacties. Injectieplaatsreacties waren in het algemeen licht tot matig in ernst; geen van deze bijwerkingen was ernstig en één van deze bijwerkingen leidde tot stopzetting van guselkumab.
In twee klinische fase III-studies bij arthritis psoriatica was t/m week 24 het aantal patiënten dat 1 of meer injectieplaatsreacties meldde laag en iets hoger in de guselkumab-groepen dan in de placebogroep; 5 (1,3%) patiënten in de guselkumab-q8w-groep, 4 (1,1%) patiënten in de guselkumab-q4w-groep en 1 (0,3%) patiënt in de placebogroep. Eén patiënt stopte met guselkumab vanwege een injectieplaatsreactie tijdens de placebogecontroleerde periode van de klinische studies bij arthritis psoriatica. In de loop van 1 jaar was het percentage patiënten dat melding maakte van 1 of meer injectieplaatsreacties 1,6% in de q8w-guselkumab-groep en 2,4% in de q4w-guselkumab-groep. Over het geheel genomen was het percentage injecties geassocieerd met injectieplaatsreacties waargenomen in klinische studies bij arthritis psoriatica, in de hele placebogecontroleerde periode gelijk aan de percentages waargenomen in de klinische studies bij psoriasis.
In de klinische fase III‑onderhoudsstudie bij colitis ulcerosa was t/m week 44 het aantal patiënten dat 1 of meer injectieplaatsreacties op guselkumab meldde 7,9% (2,5% van de injecties) in de guselkumab 200 mg subcutane q4w‑groep (guselkumab 200 mg werd toegediend als twee injecties van 100 mg in de klinische fase III‑onderhoudsstudie bij colitis ulcerosa) en geen injectieplaatsreacties in de guselkumab 100 mg subcutane q8w‑groep. De meeste injectieplaatsreacties waren licht en geen enkele was ernstig.
In klinische fase II- en III-studies bij de ziekte van Crohn was t/m week 48 het aantal patiënten dat 1 of meer injectieplaatsreacties op guselkumab meldde 4,1% (0,8% van de injecties) in de behandelgroep die guselkumab 200 mg intraveneuze inductie gevolgd door 200 mg subcutaan q4w kreeg en 1,4% van de patiënten (0,6% van de injecties) in de groep die guselkumab 200 mg intraveneuze inductie gevolgd door 100 mg subcutaan q8w kreeg. Over het geheel waren de injectieplaatsreacties licht, geen enkele was ernstig.
In een klinische fase III-studie bij de ziekte van Crohn was t/m week 48 het aantal patiënten dat 1 of meer injectieplaatsreacties op guselkumab meldde 7% (1,3% van de injecties) in de behandelgroep die guselkumab 400 mg subcutane inductie gevolgd door 200 mg subcutaan q4w kreeg en 4,3% van de patiënten (0,7% van de injecties) in de groep die guselkumab 400 mg subcutane inductie gevolgd door 100 mg subcutaan q8w kreeg. De meeste injectieplaatsreacties waren licht, geen enkele was ernstig.
Immunogeniciteit
De immunogeniciteit van guselkumab werd geëvalueerd met behulp van een gevoelige, voor het geneesmiddel tolerante immunoassay.
In samengevoegde analyses van fase II- en fase III-studies bij patiënten met psoriasis en arthritis psoriatica ontwikkelden zich in een periode van maximaal 52 weken behandeling bij 5% (n=145) van de met guselkumab behandelde patiënten antilichamen tegen het geneesmiddel. Van de patiënten bij wie zich antilichamen tegen het geneesmiddel ontwikkelden, had ongeveer 8% (n=12) antilichamen die als neutraliserend werden geclassificeerd, wat gelijkstaat aan 0,4% van alle patiënten die met guselkumab werden behandeld. In samengevoegde analyses van fase III-studies bij patiënten met psoriasis ontwikkelde ongeveer 15% van de met guselkumab behandelde patiënten antilichamen tegen het geneesmiddel in een periode van maximaal 264 weken behandeling. Van de patiënten bij wie zich antilichamen tegen het geneesmiddel ontwikkelden, had ongeveer 5% antilichamen die als neutraliserend werden geclassificeerd. Dit staat gelijk aan 0,76% van alle patiënten die met guselkumab werden behandeld. Er was geen verband tussen de vorming van antilichamen tegen het geneesmiddel en een lagere werkzaamheid of de ontwikkeling van injectieplaatsreacties.
In samengevoegde analyses van fase II‑ en fase III‑studies bij patiënten met colitis ulcerosa die werden behandeld met intraveneuze inductie gevolgd door subcutane onderhoudsbehandeling, ontwikkelde ongeveer 12% (n=58) van de patiënten die tot 56 weken met guselkumab behandeld werden antilichamen tegen het geneesmiddel. Van de patiënten bij wie zich antilichamen tegen het geneesmiddel ontwikkelden, had ongeveer 16% (n=9) antilichamen die als neutraliserend werden geclassificeerd, wat gelijkstaat aan 2% van alle patiënten die met guselkumab werden behandeld. In een fase III‑analyse bij patiënten met colitis ulcerosa die werden behandeld met subcutane inductie gevolgd door subcutane onderhoudsbehandeling, ontwikkelde tot week 24 ongeveer 9% (n=24) van de met guselkumab behandelde patiënten antilichamen tegen het geneesmiddel. Van de patiënten bij wie zich antilichamen tegen het geneesmiddel ontwikkelden, had 13% (n=3) antilichamen die als neutraliserend werden geclassificeerd, wat gelijkstaat aan 1% van de met guselkumab behandelde patiënten. Er was geen verband tussen de vorming van antilichamen tegen het geneesmiddel en een lagere werkzaamheid of de ontwikkeling van injectieplaatsreacties.
In samengevoegde analyses van fase II- en fase III-studies bij patiënten met de ziekte van Crohn die werden behandeld met intraveneuze inductie gevolgd door een subcutaan onderhoudsdoseringsschema ontwikkelden zich tot week 48 bij ongeveer 5% (n=30) van de patiënten behandeld met guselkumab antilichamen tegen het geneesmiddel. Van de patiënten bij wie zich antilichamen tegen het geneesmiddel ontwikkelden, had ongeveer 7% (n=2) antilichamen die als neutraliserende antilichamen werden geclassificeerd, wat gelijkstaat aan 0,3% van de met guselkumab behandelde patiënten. In een analyse van een fase III-studie bij patiënten met de ziekte van Crohn die waren behandeld met subcutane inductie gevolgd door een subcutaan onderhoudsdoseringsschema, ontwikkelde tot week 48 ongeveer 9% (n=24) van de met guselkumab behandelde patiënten antilichamen tegen het geneesmiddel. Van die patiënten had 13% (n=3) antilichamen die als neutraliserende antilichamen werden geclassificeerd. Dit staat gelijk aan 1% van de met guselkumab behandelde patiënten. Er was geen verband tussen de vorming van antilichamen tegen het geneesmiddel en een lagere werkzaamheid of de ontwikkeling van injectieplaatsreacties.
Pediatrische patiënten
Plaque psoriasis
De veiligheid van guselkumab werd beoordeeld in een fase III-studie met placebocontrole en actieve controle bij pediatrische patiënten met matige tot ernstige plaque psoriasis. In deze klinische studie werd de veiligheid tot 52 weken geëvalueerd bij 120 patiënten van 6 tot 17 jaar. Het veiligheidsprofiel van guselkumab toegediend via subcutane injectie met de 45 mg/0,45 ml voorgevulde pen of de 100 mg voorgevulde spuit bij pediatrische patiënten van 6 tot 17 jaar kwam overeen met het veiligheidsprofiel dat is gerapporteerd in studies bij volwassenen met plaque psoriasis (zie rubriek 4.2).
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
E-mail: adr@fagg-afmps.be
Nederland
Nederlands Bijwerkingen Centrum Lareb
Website: www.lareb.nl
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Janssen-Cilag International NV
Turnhoutseweg 30
B‑2340 Beerse
België
8. NUMMERS VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Tremfya 100 mg OnePress oplossing voor injectie in een voorgevulde pen
EU/1/17/1234/002 1 voorgevulde pen
EU/1/17/1234/003 2 voorgevulde pennen
Tremfya 100 mg PushPen oplossing voor injectie in een voorgevulde pen
EU/1/17/1234/010 1 voorgevulde pen
EU/1/17/1234/011 2 voorgevulde pennen
10. DATUM VAN HERZIENING VAN DE TEKST
18/12/2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3782646 | TREMFYA 100MG ONEPRESS OPL INJ VOORGEVULDE PEN 1 | L04AC16 | € 1989,38 | - | Ja | € 12,8 | € 8,5 |