SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
ADVATE 250 ie/5 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 500 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 1.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 1.500 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 2.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 3.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
ADVATE 250 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 250 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 50 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
ADVATE 500 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 500 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 100 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
ADVATE 1.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 1.000 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 200 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
ADVATE 1.500 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 1.500 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 300 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
ADVATE 2.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 2.000 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 400 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
ADVATE 3.000 IE/5 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 3.000 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 600 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 5 ml oplosmiddel.
De sterkte (Internationale Eenheden) wordt bepaald aan de hand van het chromogeenonderzoek van de Europese Farmacopee. De specifieke activiteit van ADVATE bedraagt ongeveer 4.520 ‑ 11.300 IE/mg proteïne.
Octocog alfa (humane stollingsfactor VIII (rDNA)) is een gezuiverd proteïne dat bestaat uit 2.332 aminozuren. Het wordt geproduceerd met DNA‑recombinatietechniek in cellen uit het ovarium van Chinese hamsters (CHO). Bereid zonder de toevoeging van (exogene) proteïnen, afgeleid van mensen of dieren, in het celcultuurproces, de zuivering of de eindformulatie.
Hulpstoffen met bekend effect
Dit geneesmiddel bevat 0,45 mmol natrium (10 mg) en 0,5 mg polysorbaat 80 per injectieflacon.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Poeder en oplosmiddel voor oplossing voor injectie.
Poeder: bros poeder met een witte tot vaalwitte kleur.
Oplosmiddel: heldere en kleurloze oplossing.
Na reconstitutie is de oplossing helder, kleurloos en vrij van vreemde deeltjes en heeft deze een pH‑waarde van 6,7 tot 7,3.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Behandeling en preventie van bloedingen bij patiënten met hemofilie A (aangeboren factor VIII‑deficiëntie). ADVATE is geïndiceerd voor alle leeftijdsgroepen.
4.2 Dosering en wijze van toediening
De behandeling moet worden gestart onder toezicht van een arts die ervaring heeft met de behandeling van hemofilie terwijl reanimatie‑apparatuur onmiddellijk beschikbaar is in geval van anafylaxie.
Behandelingscontrole
Het wordt aanbevolen om tijdens de behandeling gepaste bepaling van niveaus van factor VIII uit te voeren voor een juiste vaststelling van de toe te dienen dosis en de frequentie van herhaalde infusies. De reactie op factor VIII en daarmee de halfwaardetijd en de herstelduur kunnen per patiënt verschillen. De dosis op basis van het lichaamsgewicht moet mogelijk worden aangepast voor patiënten met ondergewicht en overgewicht. Met name in het geval van zware chirurgische interventies is nauwkeurige controle van de substitutietherapie door middel van stollingsanalyse (factor VIII-activiteit in plasma) van essentieel belang.
Dosering
De dosis en de duur van de substitutietherapie zijn afhankelijk van de ernst van de factor VIII‑deficiëntie, de plaats en de omvang van de bloeding en de klinische toestand van de patiënt.
Het aantal toegediende eenheden van factor VIII wordt uitgedrukt in Internationale Eenheden (IE), die verband houden met de actuele WHO‑concentraatstandaard voor factor VIII‑producten. De factor VIII‑activiteit in plasma wordt uitgedrukt als een percentage (ten opzichte van normaal humaan plasma) of bij voorkeur in Internationale Eenheden (ten opzichte van een Internationale Standaard voor factor VIII in plasma).
Eén Internationale Eenheid (IE) van factor VIII‑activiteit komt overeen met de hoeveelheid factor VIII in één ml normaal humaan plasma.
Behandeling op aanvraag
De berekening van de vereiste dosis factor VIII is gebaseerd op de empirische bevinding dat 1 IE factor VIII per kg lichaamsgewicht de factor VIII‑activiteit in plasma doet stijgen met 2 IE/dl. De vereiste dosis wordt met de volgende formule bepaald:
vereiste eenheden (IE) = lichaamsgewicht (kg) x gewenste stijging van factor VIII (%) x 0,5
De toe te dienen hoeveelheid en de frequentie van toediening moeten altijd zijn gebaseerd op de klinische werkzaamheid in het individuele geval. Onder bepaalde omstandigheden (bijv. bij aanwezigheid van een remmer met lage titer) kunnen hogere doses dan de met de formule berekende doses noodzakelijk zijn.
In geval van volgende type bloedingen mag de factor VIII‑activiteit niet dalen tot onder het gegeven niveau van activiteit in plasma (uitgedrukt in % ten opzichte van de normale waarde of in IE/dl) in de overeenkomstige periode. Onderstaande tabel (Tabel 1) kan dienen als leidraad voor de dosering bij bloedingen en chirurgische ingrepen.
Tabel 1. Leidraad voor de dosering bij bloedingen en chirurgische ingrepen | ||
Ernst van de bloeding/Aard van de chirurgische ingreep | Vereist niveau van factor VIII‑activiteit (% of IE/dl) | Frequentie van doses (uur)/ Therapieduur (dagen) |
Bloeding |
|
|
Beginnende gewrichtsbloeding, spierbloeding of bloeding in de mondholte. | 20 – 40 | Herhaal de injecties om de 12 tot 24 uur (8 tot 24 uur voor patiënten jonger dan 6 jaar) gedurende ten minste 1 dag, tot de bloeding stopt, (de pijn gestild is) of tot genezing van de wond. |
Meer uitgebreide gewrichtsbloeding, spierbloeding of hematoom. | 30 – 60 | Herhaal de injecties om de 12 tot 24 uur (8 tot 24 uur voor patiënten jonger dan 6 jaar) gedurende 3 tot 4 dagen of langer, tot de pijn gestild is en het acute functieverlies verdwenen is. |
Levensbedreigende bloedingen. | 60 – 100 | Herhaal de injecties om de 8 tot 24 uur (6 tot 12 uur voor patiënten jonger dan 6 jaar) tot de toestand niet langer levensbedreigend is. |
Chirurgie |
|
|
Kleine ingrepen | 30 – 60 | Om de 24 uur (12 tot 24 uur voor patiënten jonger dan 6 jaar), ten minste 1 dag, tot genezing van de wond. |
Zware ingrepen | 80 – 100 | Herhaal de injecties om de 8 tot 24 uur (6 tot 24 uur voor patiënten jonger dan 6 jaar) tot de wond voldoende genezen is. Ga vervolgens verder met de therapie gedurende ten minste 7 bijkomende dagen om de factor VIII‑activiteit tussen 30% en 60% (IE/dl) te houden. |
Preventie
Voor langetermijnpreventie van bloedingen bij patiënten met ernstige hemofilie A bedragen de gebruikelijke doses 20 tot 40 IE factor VIII per kg lichaamsgewicht met intervallen van 2 tot 3 dagen.
In sommige gevallen, met name bij jongere patiënten, kunnen kortere doseringsintervallen of hogere doses noodzakelijk zijn.
Pediatrische patiënten
De dosis bij behandeling op aanvraag is bij pediatrische patiënten (0 tot 18 jaar) niet anders dan bij volwassen patiënten. Voor profylactische behandeling van patiënten jonger dan 6 jaar worden doses van 20 tot 50 IE factor VIII per kilo lichaamsgewicht 3 tot 4 maal per week aanbevolen.
Wijze van toediening
Intraveneus gebruik. Een passende training is vereist indien de toediening niet uitgevoerd wordt door een beroepsbeoefenaar in de gezondheidszorg.
De toedieningssnelheid moet zo worden bepaald, dat de patiënt die als aangenaam ervaart, en bedraagt maximaal 10 ml/min.
Voor instructies over reconstitutie van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra‑indicaties
Overgevoeligheid voor de werkzame stoffen of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Bekende allergische reactie op muizen- of hamsterproteïnen.
4.8 Bijwerkingen
Overzicht veiligheidsprofiel
Tijdens klinisch onderzoek met ADVATE bij 418 personen met minimaal één blootstelling aan ADVATE zijn in totaal 93 bijwerkingen (ADR’s) gemeld. De bijwerkingen die het vaakst voorkwamen, waren de vorming van neutraliserende antistoffen voor factor VIII (remmers), hoofdpijn en koorts.
Overgevoeligheidsreacties of allergische reacties (zoals angio‑oedeem, branderig en stekend gevoel op de toedieningsplaats, koude rillingen, overmatig blozen, gegeneraliseerde urticaria, hoofdpijn, galbulten, hypotensie, lethargie, nausea, rusteloosheid, tachycardie, beklemd gevoel in de borst, tintelen, braken, piepende ademhaling) worden zelden waargenomen en kunnen zich in sommige gevallen ontwikkelen tot ernstige anafylaxie (waaronder shock).
Ontwikkeling van antistoffen tegen muizen‑ en/of hamsterproteïne met gerelateerde overgevoeligheidsreacties kan worden waargenomen.
Bij patiënten met hemofilie A die zijn behandeld met factor VIII, waaronder ADVATE (zie rubriek 5.1), kunnen zich neutraliserende antistoffen (remmers) vormen. Indien dergelijke remmers voorkomen, uit zich dit in een onvoldoende klinische respons. In deze gevallen wordt aangeraden contact op te nemen met een gespecialiseerd hemofiliecentrum.
Overzicht bijwerkingen in tabelvorm
Tabel 2 geeft de frequentie van bijwerkingen aan in klinisch onderzoek en afkomstig van spontane rapportering, gerangschikt volgens de systeem/orgaanklassen van de MedDRA‑gegevensbank en hun frequentie (systeem/orgaanklasse en voorkeursterm).
De frequentie is geëvalueerd op basis van de volgende conventie: zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000), niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen iedere frequentiegroep worden bijwerkingen gerangschikt naar afnemende ernst.
Tabel 2. Frequentie van bijwerkingen in klinisch onderzoek en afkomstig van spontane rapportering | ||
Standaard MedDRA‑systeem/orgaanklasse | Bijwerking | Frequentiea |
Infecties en parasitaire aandoeningen | griep | soms |
laryngitis | soms | |
Bloed‑ en lymfestelselaandoeningen | factor VIII‑remming | soms (PTP’s)b |
lymfangitis | soms | |
Immuunsysteemaandoeningen | anafylactische reactie* | niet bekend |
overgevoeligheidc* | niet bekend | |
Zenuwstelsel‑aandoeningen | hoofdpijn | vaak |
duizeligheid | soms | |
geheugenstoornis | soms | |
syncope | soms | |
trillen | soms | |
migraine | soms | |
dysgeusie | soms | |
Oogaandoeningen | oogontsteking | soms |
Hartaandoeningen | hartkloppingen | soms |
Bloedvataandoeningen | hematoom | soms |
opvliegingen | soms | |
bleekheid | soms | |
Ademhalingsstelsel‑, borstkas‑ en mediastinum‑aandoeningen | dyspneu | soms |
Maag‑darmstelsel‑aandoeningen | diarree | soms |
pijn in de bovenbuik | soms | |
misselijkheid | soms | |
braken | soms | |
Huid‑ en onderhuidaandoeningen | jeuk | soms |
huiduitslag | soms | |
overmatig zweten | soms | |
urticaria | soms | |
Algemene aandoeningen en toedieningsplaats‑stoornissen | pyrexie | vaak |
perifeer oedeem | soms | |
pijn op de borst | soms | |
borstongemak | soms | |
koude rillingen | soms | |
zich niet normaal voelen | soms | |
hematoom op punctieplaats van bloedvat | soms | |
vermoeidheid* | niet bekend | |
injectieplaatsreactie* | niet bekend | |
malaise* | niet bekend | |
Onderzoeken | verhoogd aantal monocyten | soms |
verlaagd niveau van factor VIII‑stollingsactiviteitd | soms | |
verminderde hematocrietwaarde | soms | |
abnormale laboratoriumtests | soms | |
Letsels, intoxicaties en verrichtingscomplicaties | complicatie na de verrichting | soms |
bloeding na de verrichting | soms | |
reactie op de plaats van verrichting | soms | |
a) Berekend op basis van het totale aantal patiënten die ADVATE toegediend kregen (418) in klinische onderzoeken, met uitzondering van bijwerkingen die werden geïdentificeerd tijdens het toezicht na het in de handel brengen; deze zijn voorzien van een ‘*’.
b) De frequentie is gebaseerd op onderzoeken met alle producten met factor VIII waaraan patiënten met ernstige hemofilie A deelnamen. PTP’s = eerder behandelde patiënten, PUP’s = niet eerder behandelde patiënten.
c) ADR uitgelegd in de rubriek hierna.
d) De onverwachte daling van de niveaus van factor VIII-stollingsactiviteit is waargenomen bij één patiënt tijdens de continue infusie van ADVATE na de chirurgische ingreep (dag 10 – 14 na de operatie). Gedurende deze periode was hemostase steeds behouden. Zowel de niveaus van factor VIII activiteit in plasma als de klaringssnelheden bereikten opnieuw de normale waarden op dag 15 na de operatie. De onderzoeken naar factor VIII remmers uitgevoerd na beëindiging van de continue infusie en het onderzoek waren negatief.
Beschrijving van geselecteerde bijwerkingen
ADR’s specifiek voor residuen van het productieproces
Van de 229 behandelde patiënten die beoordeeld zijn op antistoffen tegen proteïnen geproduceerd in cellen uit het ovarium van Chinese hamsters (CHO), lieten er 3 een statistisch significante opwaartse trend in titers zien, vertoonden er 4 aanhoudende pieken of voorbijgaande spikes, en vertoonde één patiënt allebei, maar had geen klinische symptomen. Van de 229 behandelde patiënten die beoordeeld zijn op antistoffen tegen muriene IgG, lieten er 10 een statistisch significante opwaartse trend zien, vertoonden er 2 een aanhoudende piek of voorbijgaande spike en vertoonde één patiënt allebei. Vier van deze patiënten meldden geïsoleerde gevallen van urticaria, jeuk, huiduitslag en licht verhoogde eosinofiele tellingen onder herhaalde blootstellingen aan het onderzoeksproduct.
Overgevoeligheidsreacties
Allergische overgevoeligheidsreacties omvatten anafylaxie en hebben zich gemanifesteerd als duizeligheid, paresthesie, huiduitslag, roodheid, aangezichtszwelling, urticaria en pruritus.
Pediatrische patiënten
De frequentie, het type en de ernst van bijwerkingen bij kinderen zijn naar verwachting hetzelfde als bij volwassenen.
Naast de ontwikkeling van remmers in eerder onbehandelde pediatrische patiënten (PUP’s) en complicaties als gevolg van de katheter, zijn er geen leeftijdsspecifieke verschillen in ADR’s opgemerkt tijdens klinisch onderzoek.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Takeda Manufacturing Austria AG
Industriestrasse 67
A‑1221 Wenen
Oostenrijk
medinfoEMEA@takeda.com
8. NUMMERS VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/03/271/001
EU/1/03/271/002
EU/1/03/271/003
EU/1/03/271/004
EU/1/03/271/005
EU/1/03/271/006
EU/1/03/271/011
EU/1/03/271/012
EU/1/03/271/013
EU/1/03/271/014
EU/1/03/271/015
EU/1/03/271/016
10. DATUM VAN HERZIENING VAN DE TEKST
05/2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau (https://www.ema.europa.eu).
SAMENVATTING VAN DE PRODUCTKENMERKEN
ADVATE 250 ie/2 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 500 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 1.000 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 1.500 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
ADVATE 250 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 250 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 125 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 2 ml oplosmiddel.
ADVATE 500 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 500 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 250 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 2 ml oplosmiddel.
ADVATE 1.000 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 1.000 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 500 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 2 ml oplosmiddel.
ADVATE 1.500 IE/2 ml, poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 1.500 IE humane stollingsfactor VIII (rDNA), octocog alfa. ADVATE bevat ongeveer 750 IE humane stollingsfactor VIII (rDNA) per ml, octocog alfa na reconstitutie met 2 ml oplosmiddel.
De sterkte (Internationale Eenheden) wordt bepaald aan de hand van het chromogeenonderzoek van de Europese Farmacopee. De specifieke activiteit van ADVATE bedraagt ongeveer 4.520 ‑ 11.300 IE/mg proteïne.
Octocog alfa (humane stollingsfactor VIII (rDNA)) is een gezuiverd proteïne dat bestaat uit 2.332 aminozuren. Het wordt geproduceerd met DNA‑recombinatietechniek in cellen uit het ovarium van Chinese hamsters (CHO). Bereid zonder de toevoeging van (exogene) proteïnen, afgeleid van mensen of dieren, in het celcultuurproces, de zuivering of de eindformulatie.
Hulpstoffen met bekend effect
Dit geneesmiddel bevat 0,45 mmol natrium (10 mg) en 0,5 mg polysorbaat 80 per injectieflacon.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
Poeder en oplosmiddel voor oplossing voor injectie.
Poeder: bros poeder met een witte tot vaalwitte kleur.
Oplosmiddel: heldere en kleurloze oplossing.
Na reconstitutie is de oplossing helder, kleurloos en vrij van vreemde deeltjes en heeft deze een pH‑waarde van 6,7 tot 7,3.
Behandeling en preventie van bloedingen bij patiënten met hemofilie A (aangeboren factor VIII‑deficiëntie). ADVATE is geïndiceerd voor alle leeftijdsgroepen.
De behandeling moet worden gestart onder toezicht van een arts die ervaring heeft met de behandeling van hemofilie terwijl reanimatie‑apparatuur onmiddellijk beschikbaar is in geval van anafylaxie.
Behandelingscontrole
Het wordt aanbevolen om tijdens de behandeling gepaste bepaling van niveaus van factor VIII uit te voeren voor een juiste vaststelling van de toe te dienen dosis en de frequentie van herhaalde infusies. De reactie op factor VIII en daarmee de halfwaardetijd en de herstelduur kunnen per patiënt verschillen. De dosis op basis van het lichaamsgewicht moet mogelijk worden aangepast voor patiënten met ondergewicht en overgewicht. Met name in het geval van zware chirurgische interventies is nauwkeurige controle van de substitutietherapie door middel van stollingsanalyse (factor VIII-activiteit in plasma) van essentieel belang.
Dosering
De dosis en de duur van de substitutietherapie zijn afhankelijk van de ernst van de factor VIII‑deficiëntie, de plaats en de omvang van de bloeding en de klinische toestand van de patiënt.
Het aantal toegediende eenheden van factor VIII wordt uitgedrukt in Internationale Eenheden (IE), die verband houden met de actuele WHO‑concentraatstandaard voor factor VIII‑producten. De factor VIII‑activiteit in plasma wordt uitgedrukt als een percentage (ten opzichte van normaal humaan plasma) of bij voorkeur in Internationale Eenheden (ten opzichte van een Internationale Standaard voor factor VIII in plasma).
Eén Internationale Eenheid (IE) van factor VIII‑activiteit komt overeen met de hoeveelheid factor VIII in één ml normaal humaan plasma.
Behandeling op aanvraag
De berekening van de vereiste dosis factor VIII is gebaseerd op de empirische bevinding dat 1 IE factor VIII per kg lichaamsgewicht de factor VIII‑activiteit in plasma doet stijgen met 2 IE/dl. De vereiste dosis wordt met de volgende formule bepaald:
vereiste eenheden (IE) = lichaamsgewicht (kg) x gewenste stijging van factor VIII (%) x 0,5
De toe te dienen hoeveelheid en de frequentie van toediening moeten altijd zijn gebaseerd op de klinische werkzaamheid in het individuele geval. Onder bepaalde omstandigheden (bijv. bij aanwezigheid van een remmer met lage titer) kunnen hogere doses dan de met de formule berekende doses noodzakelijk zijn.
In geval van volgende type bloedingen mag de factor VIII‑activiteit niet dalen tot onder het gegeven niveau van activiteit in plasma (uitgedrukt in % ten opzichte van de normale waarde of in IE/dl) in de overeenkomstige periode. Onderstaande tabel (Tabel 1) kan dienen als leidraad voor de dosering bij bloedingen en chirurgische ingrepen.
Tabel 1. Leidraad voor de dosering bij bloedingen en chirurgische ingrepen | ||
Ernst van de bloeding/Aard van de chirurgische ingreep | Vereist niveau van factor VIII‑activiteit (% of IE/dl) | Frequentie van doses (uur)/ Therapieduur (dagen) |
Bloeding |
|
|
Beginnende gewrichtsbloeding, spierbloeding of bloeding in de mondholte. | 20 – 40 | Herhaal de injecties om de 12 tot 24 uur (8 tot 24 uur voor patiënten jonger dan 6 jaar) gedurende ten minste 1 dag, tot de bloeding stopt (de pijn gestild is) of tot genezing van de wond. |
Meer uitgebreide gewrichtsbloeding, spierbloeding of hematoom | 30 – 60 | Herhaal de injecties om de 12 tot 24 uur (8 tot 24 uur voor patiënten jonger dan 6 jaar) gedurende 3 tot 4 dagen of langer, tot de pijn gestild is en het acute functieverlies verdwenen is. |
Levensbedreigende bloedingen. | 60 – 100 | Herhaal de injecties om de 8 tot 24 uur (6 tot 12 uur voor patiënten jonger dan 6 jaar) tot de toestand niet langer levensbedreigend is. |
Chirurgie |
|
|
Kleine ingrepen | 30 – 60 | Om de 24 uur (12 tot 24 uur voor patiënten jonger dan 6 jaar), ten minste 1 dag, tot genezing van de wond. |
Zware ingrepen | 80 – 100 | Herhaal de injecties om de 8 tot 24 uur (6 tot 24 uur voor patiënten jonger dan 6 jaar) tot de wond voldoende genezen is. Ga vervolgens verder met de therapie gedurende ten minste 7 bijkomende dagen om de factor VIII‑activiteit tussen 30% en 60% (IE/dl) te houden. |
Preventie
Voor langetermijnpreventie van bloedingen bij patiënten met ernstige hemofilie A bedragen de gebruikelijke doses 20 tot 40 IE factor VIII per kg lichaamsgewicht met intervallen van 2 tot 3 dagen.
In sommige gevallen, met name bij jongere patiënten, kunnen kortere doseringsintervallen of hogere doses noodzakelijk zijn.
Pediatrische patiënten
De dosis bij behandeling op aanvraag is bij pediatrische patiënten (0 tot 18 jaar) niet anders dan bij volwassen patiënten. Voor profylactische behandeling van patiënten jonger dan 6 jaar worden doses van 20 tot 50 IE factor VIII per kilo lichaamsgewicht 3 tot 4 maal per week aanbevolen.
Wijze van toediening
Intraveneus gebruik. Een passende training is vereist indien de toediening niet uitgevoerd wordt door een beroepsbeoefenaar in de gezondheidszorg.
De toedieningssnelheid moet zo worden bepaald, dat de patiënt die als aangenaam ervaart, en bedraagt maximaal 10 ml/min.
Voor instructies over reconstitutie van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
Overgevoeligheid voor de werkzame stoffen of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Bekende allergische reactie op muizen- of hamsterproteïnen.
Terugvinden herkomst
Om het terugvinden van de herkomst van biologicals te verbeteren moeten de naam en het batchnummer van het toegediende product goed geregistreerd te worden.
Overgevoeligheid
Allergische overgevoeligheidsreacties, waaronder anafylaxie, zijn gerapporteerd bij ADVATE. Het product bevat sporen van muizen‑ en hamsterproteïnen. Indien symptomen van overgevoeligheid optreden, moet patiënten worden aangeraden het gebruik van het product onmiddellijk stop te zetten en contact op te nemen met hun arts. Patiënten moeten worden geïnformeerd over de vroege tekenen van overgevoeligheidsreacties, zoals galbulten, gegeneraliseerde urticaria, beklemd gevoel op de borst, piepende ademhaling, hypotensie en anafylaxie.
In geval van shock moet de standaard medische behandeling van shock worden toegepast.
Vanwege de daling van het injectievolume van ADVATE gereconstitueerd met 2 ml gesteriliseerd water voor injecties, is er in geval van overgevoeligheidsreacties minder tijd om te reageren door de injectie te stoppen. Om deze reden is voorzichtigheid geboden bij de injectie van ADVATE gereconstitueerd in 2 ml gesteriliseerd water voor injecties, vooral bij kinderen.
Remmers
De vorming van neutraliserende antistoffen (remmers) tegen factor VIII is een bekende complicatie bij de behandeling van patiënten met hemofilie A. Deze remmers zijn doorgaans IgG‑immunoglobulinen, gericht tegen de prostollingsactiviteit van factor VIII, die aan de hand van het aangepaste onderzoek gekwantificeerd worden in Bethesda‑eenheden (BE) per ml plasma. Het risico op vorming van remmers houdt verband met de ernst van de aandoening en de blootstelling aan factor VIII, waarbij dit risico het grootst is tijdens de eerste 50 behandelingsdagen, maar het hele leven aanhoudt, hoewel het risico niet vaak voorkomt.
De klinische relevantie van de vorming van remmers is afhankelijk van de titer van de remmer, waarbij geldt dat lage titers minder risico op een onvoldoende klinische respons opleveren dan remmers met hoge titers.
In het algemeen moeten alle patiënten die met producten met stollingsfactor VIII behandeld worden nauwkeurig worden gecontroleerd en gevolgd aan de hand van relevante klinische waarnemingen en laboratoriumtests om de vorming van remmers na te gaan. Indien de verwachte niveaus van factor VIII‑activiteit in plasma niet verkregen worden of indien de bloeding niet onder controle gehouden wordt met een aangepaste dosis, moet een onderzoek worden uitgevoerd om na te gaan of er een factor VIII‑remmer aanwezig is. Bij patiënten met een hoge spiegel aan remmers is het mogelijk dat de factor VIII‑therapie niet doeltreffend is en moeten alternatieve therapieën worden overwogen. De behandeling van dergelijke patiënten moet worden uitgevoerd door artsen die ervaring hebben met de behandeling van hemofilie en met factor VIII‑remmers.
Verkeerde toediening van ADVATE
Bij toediening van ADVATE gereconstitueerd met 2 ml gesteriliseerd water voor injecties, kan verkeerde toediening (intra‑arterieel of paraveneus) leiden tot milde, kortdurende reacties op de injectieplaats, zoals blauwe plekken en erytheem.
Cardiovasculaire voorvallen
Bij patiënten met bestaande cardiovasculaire risicofactoren kan substitutietherapie met factor VIII leiden tot een verhoging van het cardiovasculaire risico.
Complicaties als gevolg van de katheter
Indien een instrument voor veneuze toegang (CVAD) is vereist, moet rekening worden gehouden met het risico van complicaties als gevolg van het CVAD waaronder lokale infecties, bacteriëmie en trombose op de katheterlocatie.
Overwegingen in verband met hulpstoffen
Natrium
Dit middel bevat 10 mg natrium per injectieflacon, overeenkomend met 0,5% van de door de WHO aanbevolen maximale dagelijkse inname van 2 g voor een volwassene.
Het wordt sterk aanbevolen de naam en het batchnummer van het product te noteren telkens wanneer ADVATE toegediend wordt, zodat een verband kan worden gelegd tussen de patiënt enerzijds en de geneesmiddelbatch anderzijds.
Pediatrische patiënten
De vermelde waarschuwingen en voorzorgen bij gebruik gelden voor zowel volwassenen als kinderen.
Er zijn geen interacties van humane stollingsfactor VIII (rDNA)-producten met andere geneesmiddelproducten gemeld.
Er is geen reproductieonderzoek bij dieren uitgevoerd met factor VIII. Vanwege het zeldzaam voorkomen van hemofilie A bij vrouwen bestaat er geen ervaring met het gebruik van factor VIII tijdens zwangerschap en borstvoeding. Daarom mag factor VIII alleen worden toegediend tijdens zwangerschap en borstvoeding indien het gebruik duidelijk geïndiceerd is.
ADVATE heeft geen of een verwaarloosbare invloed op de rijvaardigheid en op het vermogen om machines te bedienen.
Overzicht veiligheidsprofiel
Tijdens klinisch onderzoek met ADVATE bij 418 personen met minimaal één blootstelling aan ADVATE zijn in totaal 93 bijwerkingen (ADR’s) gemeld. De bijwerkingen die het vaakst voorkwamen, waren de vorming van neutraliserende antistoffen voor factor VIII (remmers), hoofdpijn en koorts.
Overgevoeligheidsreacties of allergische reacties (zoals angio‑oedeem, branderig en stekend gevoel op de toedieningsplaats, koude rillingen, overmatig blozen, gegeneraliseerde urticaria, hoofdpijn, galbulten, hypotensie, lethargie, nausea, rusteloosheid, tachycardie, beklemd gevoel in de borst, tintelen, braken, piepende ademhaling) worden zelden waargenomen en kunnen zich in sommige gevallen ontwikkelen tot ernstige anafylaxie (waaronder shock).
Ontwikkeling van antistoffen tegen muizen‑ en/of hamsterproteïne met gerelateerde overgevoeligheidsreacties kan worden waargenomen.
Bij patiënten met hemofilie A die zijn behandeld met factor VIII, waaronder ADVATE (zie rubriek 5.1), kunnen zich neutraliserende antistoffen (remmers) vormen. Indien dergelijke remmers voorkomen, uit zich dit in een onvoldoende klinische respons. In deze gevallen wordt aangeraden contact op te nemen met een gespecialiseerd hemofiliecentrum.
Overzicht bijwerkingen in tabelvorm
Tabel 2 geeft de frequentie van bijwerkingen aan in klinisch onderzoek en afkomstig van spontane rapportering, gerangschikt volgens de systeem/orgaanklassen van de MedDRA‑gegevensbank en hun frequentie (systeem/orgaanklasse en voorkeursterm).
De frequentie is geëvalueerd op basis van de volgende conventie: zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000), niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen iedere frequentiegroep worden bijwerkingen gerangschikt naar afnemende ernst.
Tabel 2. Frequentie van bijwerkingen in klinisch onderzoek en afkomstig van spontane rapportering | ||
Standaard MedDRA‑systeem/orgaanklasse | Bijwerking | Frequentiea |
Infecties en parasitaire aandoeningen | griep | soms |
laryngitis | soms | |
Bloed‑ en lymfestelselaandoeningen | factor VIII‑remming | soms (PTP’s)b |
lymfangitis | soms | |
Immuunsysteemaandoeningen | anafylactische reactie* | niet bekend |
overgevoeligheidc* | niet bekend | |
Zenuwstelsel‑aandoeningen | hoofdpijn | vaak |
duizeligheid | soms | |
geheugenstoornis | soms | |
syncope | soms | |
trillen | soms | |
migraine | soms | |
dysgeusie | soms | |
Oogaandoeningen | oogontsteking | soms |
Hartaandoeningen | hartkloppingen | soms |
Bloedvataandoeningen | hematoom | soms |
opvliegingen | soms | |
bleekheid | soms | |
Ademhalingsstelsel‑, borstkas‑ en mediastinum‑aandoeningen | dyspneu | soms |
Maag‑darmstelsel‑aandoeningen | diarree | soms |
pijn in de bovenbuik | soms | |
misselijkheid | soms | |
braken | soms | |
Huid‑ en onderhuidaandoeningen | jeuk | soms |
huiduitslag | soms | |
overmatig zweten | soms | |
urticaria | soms | |
Algemene aandoeningen en toedieningsplaats‑stoornissen | pyrexie | vaak |
perifeer oedeem | soms | |
pijn op de borst | soms | |
borstongemak | soms | |
koude rillingen | soms | |
zich niet normaal voelen | soms | |
hematoom op punctieplaats van bloedvat | soms | |
vermoeidheid* | niet bekend | |
injectieplaatsreactie* | niet bekend | |
malaise* | niet bekend | |
Onderzoeken | verhoogd aantal monocyten | soms |
verlaagd niveau van factor VIII‑stollingsactiviteitd | soms | |
verminderde hematocrietwaarde | soms | |
abnormale laboratoriumtests | soms | |
Letsels, intoxicaties en verrichtingscomplicaties | complicatie na de verrichting | soms |
bloeding na de verrichting | soms | |
reactie op de plaats van verrichting | soms | |
a) Berekend op basis van het totale aantal patiënten die ADVATE toegediend kregen (418) in klinische onderzoeken, met uitzondering van bijwerkingen die werden geïdentificeerd tijdens het toezicht na het in de handel brengen; deze zijn voorzien van een ‘*’.
b) De frequentie is gebaseerd op onderzoeken met alle producten met factor VIII waaraan patiënten met ernstige hemofilie A deelnamen. PTP’s = eerder behandelde patiënten, PUP’s = niet eerder behandelde patiënten.
c) ADR uitgelegd in de rubriek hierna.
d) De onverwachte daling van de niveaus van factor VIII-stollingsactiviteit is waargenomen bij één patiënt tijdens de continue infusie van ADVATE na de chirurgische ingreep (dag 10 – 14 na de operatie). Gedurende deze periode was hemostase steeds behouden. Zowel de niveaus van factor VIII activiteit in plasma als de klaringssnelheden bereikten opnieuw de normale waarden op dag 15 na de operatie. De onderzoeken naar factor VIII remmers uitgevoerd na beëindiging van de continue infusie en het onderzoek waren negatief.
Beschrijving van geselecteerde bijwerkingen
ADR’s specifiek voor residuen van het productieproces
Van de 229 behandelde patiënten die beoordeeld zijn op antistoffen tegen proteïnen geproduceerd in cellen uit het ovarium van Chinese hamsters (CHO), lieten er 3 een statistisch significante opwaartse trend in titers zien, vertoonden er 4 aanhoudende pieken of voorbijgaande spikes, en vertoonde één patiënt allebei, maar had geen klinische symptomen. Van de 229 behandelde patiënten die beoordeeld zijn op antistoffen tegen muriene IgG, lieten er 10 een statistisch significante opwaartse trend zien, vertoonden er 2 een aanhoudende piek of voorbijgaande spike en vertoonde één patiënt allebei. Vier van deze patiënten meldden geïsoleerde gevallen van urticaria, jeuk, huiduitslag en licht verhoogde eosinofiele tellingen onder herhaalde blootstellingen aan het onderzoeksproduct.
Overgevoeligheidsreacties
Allergische overgevoeligheidsreacties omvatten anafylaxie en hebben zich gemanifesteerd als duizeligheid, paresthesie, huiduitslag, roodheid, aangezichtszwelling, urticaria en pruritus.
Pediatrische patiënten
De frequentie, het type en de ernst van bijwerkingen bij kinderen zijn naar verwachting hetzelfde als bij volwassenen.
Naast de ontwikkeling van remmers in eerder onbehandelde pediatrische patiënten (PUP’s) en complicaties als gevolg van de katheter, zijn er geen leeftijdsspecifieke verschillen in ADR’s opgemerkt tijdens klinisch onderzoek.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
Er zijn geen symptomen van overdosering gemeld met recombinante stollingsfactor VIII.
Farmacotherapeutische categorie: antihemorragische producten, bloedstollingsfactor VIII.
ATC‑code: B02BD02.
Werkingsmechanisme
ADVATE bevat recombinante stollingsfactor VIII (octocog alfa), een glycoproteïne die biologisch gelijkwaardig is aan de factor VIII-glycoproteïne in humaan plasma. Octocog alfa is een glycoproteïne die bestaat uit 2.332-aminozuren met een moleculaire massa van ongeveer 280 kD.
Het factor VIII‑/von Willebrandfactorcomplex bestaat uit twee moleculen (factor VIII en von Willebrandfactor) met verschillende fysiologische functies. Na infusie bij een patiënt met hemofilie bindt factor VIII aan endogene von Willebrandfactor in het bloed van de patiënt. Geactiveerde factor VIII fungeert als cofactor voor geactiveerde factor IX, waardoor de omzetting van factor X naar geactiveerde factor X wordt versneld. Geactiveerde factor X zet protrombine om in trombine. Vervolgens zet trombine fibrinogeen om in fibrine, waarna een stolsel kan worden gevormd. Hemofilie A is een geslachtsgebonden erfelijke stollingsstoornis die wordt veroorzaakt door verlaagde niveaus factor VIII:C en die leidt tot forse bloeding in gewrichten, spieren of inwendige organen, spontaan of als gevolg van trauma na een ongeval of chirurgische ingreep. Door substitutietherapie worden de niveaus van factor VIII in het plasma verhoogd, waardoor de factordeficiëntie en de verhoogde bloedingsneiging tijdelijk worden gecorrigeerd.
Het bloedingscijfer op jaarbasis (ABR) is niet vergelijkbaar voor verschillende factorconcentraten en verschillende klinische onderzoeken.
Klinische werkzaamheid en veiligheid
Gegevens over immuuntolerantie‑inductie (ITI) bij patiënten met remmers zijn verzameld. In een subonderzoek van PUP‑onderzoek 060103 zijn ITI‑behandelingen bij 11 PUP’s gedocumenteerd. Retrospectief dossieronderzoek is uitgevoerd onder 30 pediatrische patiënten m.b.t. ITI (in onderzoek 060703). In een niet-interventioneel prospectief registratieonderzoek (PASS-INT-004) werd ITI gedocumenteerd bij 44 pediatrische en volwassen patiënten, van wie er 36 ITI-therapie voltooiden. Uit de gegevens blijkt dat immuuntolerantie kan worden bereikt.
In onderzoek 060201 zijn twee lange‑termijn profylaxeschema’s van 53 eerder behandelde patiënten vergeleken: een individueel, farmacokinetisch geleid doseringsschema (binnen een bereik van 20 tot 80 IE factor VIII per kilo lichaamsgewicht met intervallen van 72 ± 6 uur, n=23) en een standaard profylactische dosering (20 tot 40 IE/kg om de 48 ± 6 uur, n=30). Het farmacokinetisch geleide doseringsschema was (volgens een specifieke formule) gericht op handhaving van factor VIII dalspiegels ≥ 1% bij het interdoseringsinterval van 72 uur. Gegevens uit dit onderzoek tonen aan dat de twee profylactische doseringsschema's vergelijkbaar zijn in termen van reductie van het bloedingspercentage.
Pediatrische patiënten
Het Europees Geneesmiddelenbureau heeft besloten af te zien van de verplichting voor de fabrikant om de resultaten in te dienen van onderzoek met ADVATE in alle subgroepen van pediatrische patiënten met hemofilie A (aangeboren factor VIII‑deficiëntie) in “Immuuntolerantie‑inductie (ITI) bij patiënten met hemofilie A (congenitale factor VIII‑deficiëntie) die factor VIII‑remmers hebben gevormd” en “Behandeling en preventie van bloeden bij patiënten met hemofilie A (congenitale factor VIII‑deficiëntie)” (zie rubriek 4.2 voor informatie over pediatrisch gebruik).
Alle farmacokinetisch onderzoek met ADVATE is uitgevoerd bij eerder behandelde patiënten met ernstige tot matig ernstige hemofilie A (factor VIII bij aanvang ≤ 2%). De plasmamonsters zijn geanalyseerd in een centraal laboratorium met behulp van een eenfasig stollingsonderzoek.
De farmacokinetische (PK-)gegevens afkomstig van in totaal 195 patiënten met ernstige hemofilie A (factor VIII bij aanvang < 1%) zijn opgenomen in de farmacokinetische analyseset volgens protocol. Categorieën van deze analysen voor zuigelingen (1 maand tot <2 jaar), kinderen (2 tot <5 jaar), oudere kinderen (5 tot <12 jaar), adolescenten (12 tot <18 jaar) en volwassenen (18 jaar en ouder) zijn gebruikt voor samenvatting van de farmacokinetische parameters, waarbij leeftijd is gedefinieerd als leeftijd ten tijde van de farmacokinetische infusie.
Tabel 3. Overzicht van farmacokinetische parameters voor ADVATE per leeftijdsgroep met ernstige hemofilie A (factor VIII bij aanvang < 1%) | |||||
Parameter (gemiddelde ± standaarddeviatie) | Zuigelingen | Kinderen | Oudere kinderen | Adolescenten | Volwassenen |
Totaal AUC (IE*·h/dl) | 1.362,1 ± 311,8 | 1.180,0 ± 432,7 | 1.506,6 ± 530,0 | 1.317,1 ± 438,6 | 1.538,5 ± 519,1 |
Aangepaste incrementele recovery bij Cmax (IE/dl per IE/kg)a | 2,2 ± 0,6 | 1,8 ± 0,4 | 2,0 ± 0,5 | 2,1 ± 0,6 | 2,2 ± 0,6 |
Halfwaardetijd (h) | 9,0 ± 1,5 | 9,6 ± 1,7 | 11,8 ± 3,8 | 12,1 ± 3,2 | 12,9 ± 4,3 |
Maximale plasmaconcentratie na infusie (IE/dl) | 110,5 ± 30,2 | 90,8 ± 19,1 | 100,5 ± 25,6 | 107,6 ± 27,6 | 111,3 ± 27,1 |
Gemiddelde verblijfsduur (h) | 11,0 ± 2,8 | 12,0 ± 2,7 | 15,1 ± 4,7 | 15,0 ± 5,0 | 16,2 ± 6,1 |
Distributievolume bij steady state (dl/kg) | 0,4 ± 0,1 | 0,5 ± 0,1 | 0,5 ± 0,2 | 0,6 ± 0,2 | 0,5 ± 0,2 |
Klaring (ml/kg*h) | 3,9 ± 0,9 | 4,8 ± 1,5 | 3,8 ± 1,5 | 4,1 ± 1,0 | 3,6 ± 1,2 |
a Berekend als (Cmax ‑ factor VIII bij aanvang) gedeeld door de dosis in IU/kg, waarbij Cmax de maximale post‑infusie factor VIII‑waarde is.
Pediatrische patiënten
De veiligheid en hemostatische werkzaamheid van ADVATE bij de pediatrische populatie zijn vergelijkbaar met die bij volwassen patiënten. De aangepaste recovery en terminale halfwaardetijd (t½) lagen ongeveer 20% lager bij jonge kinderen (jonger dan 6 jaar) dan bij volwassenen, wat deels te wijten kan zijn aan het bekende hogere plasmavolume per kilogram lichaamsgewicht bij jongere patiënten.
Momenteel zijn er geen farmacokinetische gegevens beschikbaar over het gebruik van ADVATE bij niet eerder behandelde patiënten.
Niet‑klinische gegevens duiden niet op een speciaal risico voor mensen. Deze gegevens zijn afkomstig van conventioneel onderzoek op het gebied van veiligheidsfarmacologie, acute toxicologie, toxiciteit bij herhaalde dosering, lokale toxiciteit en genotoxiciteit.
Uit een lokaal tolerantieonderzoek bij konijnen is gebleken dat ADVATE gereconstitueerd met 2 ml gesteriliseerd water voor injectie goed wordt verdragen na intraveneuze toediening. Lichte voorbijgaande roodheid op de toedieningsplaats werd waargenomen na intra‑arteriële toediening en na paraveneuze toediening. Er zijn echter geen samenhangende histopathologische veranderingen waargenomen, wat wijst op de voorbijgaande aard van deze bevindingen.
Poeder
Mannitol (E421)
natriumchloride
histidine
trehalose
calciumchloride (E509)
trometamol
polysorbaat 80 (E433)
glutathion (gereduceerd)
Oplosmiddel
gesteriliseerd water voor injecties
Bij gebrek aan onderzoek naar onverenigbaarheden, mag dit geneesmiddel niet met andere geneesmiddelen gemengd worden.
Ongeopende injectieflacon
2 jaar.
Tijdens de houdbaarheidsperiode mag het product gedurende één periode van maximaal 6 maanden worden bewaard bij kamertemperatuur (maximaal 25°C). Het einde van de bewaarperiode van 6 maanden bij kamertemperatuur moet worden genoteerd op de buitenverpakking van het product. Aan het einde van deze periode moet het product worden gebruikt of weggegooid. Het product mag niet opnieuw in de koelkast worden bewaard.
Na reconstitutie
Vanuit microbiologisch standpunt moet het product na reconstitutie onmiddellijk worden gebruikt.
Er is echter een chemische en fysische stabiliteit na opening van de verpakking aangetoond gedurende 3 uur bij 25°C.
Bewaren in de koelkast (2°C – 8°C).
Niet in de vriezer bewaren.
ADVATE met BAXJECT II: de injectieflacon met het product in de buitenverpakking bewaren ter bescherming tegen licht.
ADVATE in BAXJECT III: de verzegelde blisterverpakking in de buitenverpakking bewaren ter bescherming tegen licht.
Voor de bewaarcondities van het geneesmiddel na reconstitutie, zie rubriek 6.3.
De injectieflacon met het poeder en de injectieflacon met 2 ml oplosmiddel zijn vervaardigd van type I‑glas en afgesloten met een stop van chlorobutyl- of broombutylrubber. Het product wordt in een van de volgende configuraties geleverd:
- ADVATE met BAXJECT II: elke verpakking bevat een injectieflacon met poeder, een injectieflacon met 2 ml oplosmiddel en een hulpmiddel voor reconstitutie (BAXJECT II).
- ADVATE in BAXJECT III: elke verpakking bevat een gebruiksklaar BAXJECT III‑hulpmiddel in een verzegelde blisterverpakking (de injectieflacon met poeder en de injectieflacon met 2 ml oplosmiddel zijn bevestigd op het hulpmiddel voor reconstitutie).
Na reconstitutie van het gevriesdroogde product moet ADVATE intraveneus worden toegediend.
Het gereconstitueerde geneesmiddel moet visueel voorafgaand aan de toediening worden geïnspecteerd op de aanwezigheid van deeltjes en verkleuring. De oplossing moet helder en kleurloos zijn en mag geen vreemde deeltjes bevatten. Gebruik geen oplossingen die troebel zijn of deeltjes bevatten.
- Voor de toediening moet een luerlockspuit worden gebruikt.
- Binnen 3 uur na reconstitutie gebruiken.
- Het preparaat niet in de koelkast bewaren na reconstitutie.
- Al het ongebruikte geneesmiddel of afvalmateriaal dient te worden vernietigd overeenkomstig lokale voorschriften.
Reconstitutie met BAXJECT II
- Voor reconstitutie uitsluitend het gesteriliseerde water voor injecties en het hulpmiddel voor reconstitutie gebruiken die beide in de verpakking worden meegeleverd.
- Niet gebruiken indien de BAXJECT II, de steriele barrière of de verpakking beschadigd is of tekenen van beschadiging vertoont.
- Een aseptische techniek moet worden toegepast.
1. Indien het product nog steeds in de koelkast bewaard wordt, neem dan zowel de injectieflacon met ADVATE‑poeder als de injectieflacon met oplosmiddel uit de koelkast en laat ze op kamertemperatuur (tussen 15°C en 25°C) komen.
2. Was uw handen grondig met zeep en warm water.
3. Verwijder het kapje van de injectieflacon met poeder en de injectieflacon met oplosmiddel.
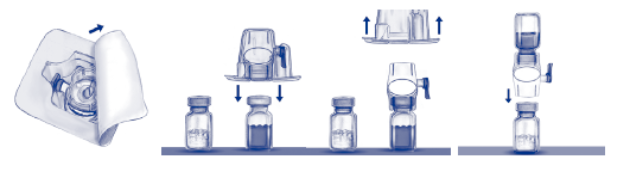
4. Reinig de stoppen met de alcoholdoekjes. Plaats de injectieflacons op een vlakke en propere ondergrond.
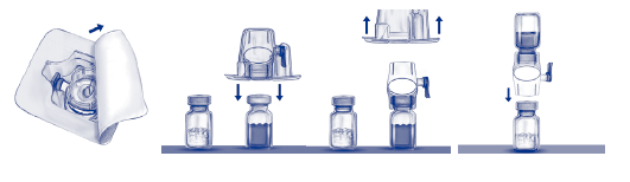
5. Verwijder de papieren beschermfolie van de verpakking zonder de binnenzijde aan te raken om de verpakking van de BAXJECT II te openen (Fig. A). Neem de BAXJECT II niet uit de verpakking. Niet gebruiken indien de BAXJECT II, de steriele barrière of de verpakking beschadigd is of tekenen van beschadiging vertoont.
6. Draai de verpakking om en druk de doorzichtige plastic spike door de stop van de injectieflacon met oplosmiddel. Houd de rand van de verpakking vast en verwijder de verpakking van de BAXJECT II (Fig. B). Laat het blauwe kapje op de BAXJECT II zitten.
7. Gebruik voor de reconstitutie uitsluitend het gesteriliseerde water voor injecties en het hulpmiddel die beide bij het product bijgeleverd zijn. De BAXJECT II is bevestigd op de injectieflacon met oplosmiddel. Draai het systeem om zodat de injectieflacon met oplosmiddel zich bovenaan bevindt. Druk de witte plastic spike door de stop van de injectieflacon met het ADVATE‑poeder. Door het vacuüm wordt het oplosmiddel opgezogen in de injectieflacon met het ADVATE‑poeder (Fig. C).
8. Zwenk voorzichtig de injectieflacon tot alle materiaal opgelost is. Controleer of het ADVATE‑poeder volledig opgelost is. Indien dat niet het geval is, kan niet de volledige gereconstitueerde oplossing de filter van het hulpmiddel passeren. Het product lost snel op (doorgaans na minder dan 1 minuut). Na reconstitutie moet de oplossing helder en kleurloos zijn en mag het geen vreemde deeltjes bevatten.
Fig. a Fig. b Fig. c
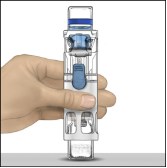
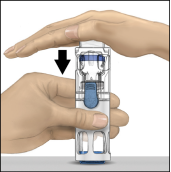
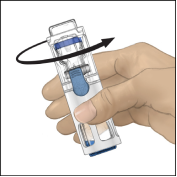
Reconstitutie met BAXJECT III
Niet gebruiken als de dop van de blisterverpakking niet volledig is verzegeld.
1. Als het product nog in de koelkast is opgeslagen, neemt u de verzegelde blisterverpakking (bevat injectieflacons met poeder en oplosmiddel, bevestigd op het hulpmiddel voor reconstitutie) uit de koelkast en laat u deze op kamertemperatuur komen (tussen 15 °C en 25 °C).
2. Was grondig uw handen met warm water en zeep.
3. Open de ADVATE‑verpakking door de dop te verwijderen. Neem BAXJECT III uit de blisterverpakking.
4. Leg de ADVATE op een vlakke ondergrond met de injectieflacon met oplosmiddel bovenop (Fig. 1). De injectieflacon met het oplosmiddel is voorzien van een blauwe streep. Verwijder de blauwe dop pas wanneer dit in een latere stap wordt gevraagd.
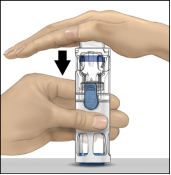
5. Houd ADVATE in BAXJECT III met één hand vast en druk met de andere hand stevig op de injectieflacon met het oplosmiddel, totdat het hulpmiddel geheel is gecollabeerd en het oplosmiddel in de ADVATE‑injectieflacon stroomt (Fig. 2). Kantel het hulpmiddel niet totdat de injectieflacon leeg is.
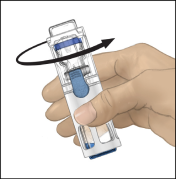
6. Controleer of alle oplosmiddel is overgebracht. Zwenk de injectieflacon voorzichtig totdat alle materiaal is opgelost (Fig. 3). Controleer of de ADVATE‑poeder volledig is opgelost omdat anders niet alle gereconstitueerde oplossing door het filter van het hulpmiddel zal stromen. Het product lost snel op (meestal in minder dan 1 minuut). Na reconstitutie moet de oplossing helder, kleurloos en vrij van vreemde deeltjes zijn.
Fig. 1 | Fig. 2 | Fig. 3 |
|
|
|
Toediening
Een aseptische techniek moet worden toegepast.
Vóór de toediening moeten parenterale geneesmiddelen worden gecontroleerd op de aanwezigheid van deeltjes, indien de oplossing en de verpakking dat mogelijk maken. Uitsluitend een heldere en kleurloze oplossing mag worden gebruikt.
1. Verwijder het blauwe kapje van de BAXJECT II / BAXJECT III. Geen lucht opzuigen in de spuit. Sluit de spuit aan op de BAXJECT II / BAXJECT III.
2. Draai het systeem om (de injectieflacon met gereconstitueerde oplossing moet zich bovenaan bevinden). Trek de zuiger langzaam achteruit om de gereconstitueerde oplossing in de spuit op te zuigen.
3. Koppel de spuit los van de BAXJECT II / BAXJECT III.
4. Bevestig een vleugelnaald op de spuit. Injecteer intraveneus. De oplossing moet langzaam worden toegediend met een snelheid van maximaal 10 ml/min die de patiënt als aangenaam ervaart. Vóór en tijdens de toediening van ADVATE moet de polsslag worden bepaald. Indien er een significante stijging optreedt, volstaat doorgaans een verlaging van de toedieningssnelheid of een tijdelijke onderbreking van de injectie om de symptomen onmiddellijk te laten verdwijnen (zie rubrieken 4.4 en 4.8).
Takeda Manufacturing Austria AG, Industriestrasse 67, A‑1221 Wenen, Oostenrijk
medinfoEMEA@takeda.com
EU/1/03/271/007
EU/1/03/271/008
EU/1/03/271/009
EU/1/03/271/010
EU/1/03/271/017
EU/1/03/271/018
EU/1/03/271/019
EU/1/03/271/020
Datum van eerste verlening van de vergunning: 2 maart 2004.
Datum van laatste verlenging: 20 december 2013.
10. DATUM VAN HERZIENING VAN DE TEKST
05/2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau (https://www.ema.europa.eu).
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 2955342 | ADVATE 2000UI PULV+SOLV SOL INJ 5 ML(400IU/ML)+KIT | B02BD02 | € 1327,95 | - | Ja | - | - |
| 2955359 | ADVATE 3000UI PULV+SOLV SOL INJ 5 ML(600IU/ML)+KIT | B02BD02 | € 1987,22 | - | Ja | - | - |
| 3342870 | ADVATE 250 UI PULV+SOLV SOL INJ 2 ML(125IU/ML)+KIT | B02BD02 | € 162,71 | - | Ja | € 2 | € 1 |
| 3342888 | ADVATE 500 UI PULV+SOLV SOL INJ 2 ML(250IU/ML)+KIT | B02BD02 | € 314,45 | - | Ja | € 2 | € 1 |
| 3342896 | ADVATE 1000UI PULV+SOLV SOL INJ 2 ML(500IU/ML)+KIT | B02BD02 | € 617,95 | - | Ja | € 2 | € 1 |
| 3342904 | ADVATE 1500UI PULV+SOLV SOL INJ 2 ML(750IU/ML)+KIT | B02BD02 | € 1003,26 | - | Ja | € 2 | € 1 |
| 3342912 | ADVATE 2000UI PULV+SOLV SOL INJ 5 ML(400IU/ML)+KIT | B02BD02 | € 1269,09 | - | Ja | € 2 | € 1 |
| 3342920 | ADVATE 3000UI PULV+SOLV SOL INJ 5 ML(600IU/ML)+KIT | B02BD02 | € 1952,9 | - | Ja | € 2 | € 1 |