1. NAAM VAN HET GENEESMIDDEL
Tecfidera 120 mg maagsapresistente capsules, hard.
Tecfidera 240 mg maagsapresistente capsules, hard.
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Tecfidera 120 mg maagsapresistente capsules, hard
Elke maagsapresistente capsule, hard bevat 120 mg dimethylfumaraat.
Tecfidera 240 mg maagsapresistente capsules, hard
Elke maagsapresistente capsule, hard bevat 240 mg dimethylfumaraat.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Maagsapresistente capsule, hard
Tecfidera 120 mg maagsapresistente capsules, hard
Groen‑witte maagsapresistente capsules, hard, maat 0, bedrukt met ‘BG‑12 120 mg’ die microtabletten bevatten.
Tecfidera 240 mg maagsapresistente capsules, hard
Groene maagsapresistente capsules, hard, maat 0, bedrukt met ‘BG‑12 240 mg’ die microtabletten bevatten.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Tecfidera is geïndiceerd voor de behandeling van volwassen en pediatrische patiënten van 13 jaar en ouder met relapsing‑remitting multipele sclerose (RRMS).
4.2 Dosering en wijze van toediening
De behandeling dient te worden geïnitieerd onder toezicht van een arts met ervaring in het behandelen van multipele sclerose.
Dosering
De startdosering is 120 mg tweemaal per dag. Na 7 dagen dient de dosis te worden verhoogd tot de aanbevolen onderhoudsdosis van 240 mg tweemaal per dag (zie rubriek 4.4).
Als een patiënt een dosis heeft overgeslagen, mag geen dubbele dosis worden ingenomen. De patiënt mag de overgeslagen dosis alleen maar innemen als er 4 uur tussen de doses is in gelaten. Anders moet de patiënt wachten tot de volgende geplande dosis.
Een tijdelijke verlaging van de dosis tot 120 mg tweemaal per dag kan het optreden van flushing en maag‑darmbijwerkingen verminderen. De aanbevolen onderhoudsdosis van 240 mg tweemaal per dag dient binnen 1 maand te worden hervat.
Tecfidera dient met voedsel te worden ingenomen (zie rubriek 5.2).Voor patiënten die flushing of maag‑darmbijwerkingen ondervinden, mag Tecfidera met voedsel worden ingenomen om de verdraagbaarheid te verbeteren (zie rubriek 4.4, 4.5 en 4.8).
Speciale populaties
Ouderen
In klinische studies met Tecfidera was er beperkte blootstelling aan patiënten van 55 jaar of ouder, en was het aantal deelnemende patiënten van 65 jaar of ouder te laag om te bepalen of deze anders reageren dan jongere patiënten (zie rubriek 5.2). Op basis van het werkingsmechanisme van de werkzame stof zijn er geen theoretische redenen om de dosis voor ouderen aan te passen.
Nier‑ en leverfunctiestoornissen
Tecfidera is niet onderzocht in patiënten met nier‑ of leverfunctiestoornissen. Op basis van klinische farmacologische studies is geen aanpassing van de dosis vereist (zie rubriek 5.2). Bij het behandelen van patiënten met ernstige nier‑ of leverfunctiestoornissen moet met voorzichtigheid worden gehandeld (zie rubriek 4.4).
Pediatrische patiënten
De dosering is dezelfde bij volwassenen en bij pediatrische patiënten van 13 jaar en ouder.
Er zijn beperkte gegevens beschikbaar voor kinderen tussen 10–12 jaar. De momenteel beschikbare gegevens worden beschreven in rubriek 4.8 en 5.1, maar er kan geen doseringsadvies worden gegeven.
De veiligheid en werkzaamheid van Tecfidera bij kinderen jonger dan 10 jaar zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Voor oraal gebruik.
De capsule moet in zijn geheel worden doorgeslikt. De capsule of diens inhoud mag niet worden fijngemaakt, verdeeld, opgelost, gezogen of gekauwd, aangezien de maagzuurbestendige omhulling van de microtabletten het irriteren van het maagdarmstelsel voorkomt.
4.3 Contra‑indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Vermoede of bevestigde progressieve multifocale leukencefalopathie (PML).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest voorkomende bijwerkingen zijn flushing (35%) en maag‑darmbijwerkingen (d.w.z. diarree (14%), misselijkheid (12%), buikpijn (10%), pijn in de bovenbuik (10%)). Flushing en maag‑darmbijwerkingen treden vaak op aan het begin van de behandeling (voornamelijk gedurende de eerste maand), en bij patiënten die flushing en maag‑darmbijwerkingen ervaren, kunnen deze voorvallen zo nu en dan blijven optreden gedurende de gehele behandeling met Tecfidera. De meest frequent gemelde bijwerkingen die tot stopzetting van de behandeling leidden, zijn flushing (3%) en maag‑darmbijwerkingen (4%).
In fase 2 en 3 placebogecontroleerde en ongecontroleerde klinische onderzoeken hebben in totaal 2.513 patiënten Tecfidera gekregen gedurende perioden van maximaal 12 jaar met een algehele blootstelling die equivalent was aan 11.318 persoonsjaren. In totaal hebben 1.169 patiënten ten minste 5 jaar en 426 patiënten ten minste 10 jaar behandeling met Tecfidera gekregen. De ervaringen uit ongecontroleerde klinische onderzoeken zijn consistent met de ervaringen uit placebogecontroleerde klinische onderzoeken.
Lijst van bijwerkingen in tabelvorm
Bijwerkingen die voortkomen uit klinische studies, veiligheidsstudies na toelating en spontane meldingen zijn in onderstaande tabel vermeld.
De bijwerkingen zijn gerangschikt naar systeem/orgaanklasse volgens de MedDRA‑gegevensbank. De frequentie van onderstaande bijwerkingen wordt als volgt aangeduid:
- Zeer vaak ( 1/10)
- Vaak ( 1/100, < 1/10)
- Soms ( 1/1.000, < 1/100)
- Zelden ( 1/10.000, < 1/1.000)
- Zeer zelden (< 1/10.000)
- Niet bekend (de frequentie kan met de beschikbare gegevens niet worden bepaald)
MedDRA- systeem/orgaanklasse | Bijwerking | Frequentiecategorie |
Infecties en parasitaire aandoeningen | Gastro‑enteritis | Vaak |
Progressieve multifocale leuko‑encefalopathie (PML) | Niet bekend | |
Herpes zoster | Niet bekend | |
Bloed‑ en lymfestelselaandoeningen | Lymfopenie | Vaak |
Leukopenie | Vaak | |
Trombocytopenie | Soms | |
Immuunsysteemaandoeningen | Overgevoeligheid | Soms |
Anafylaxie | Niet bekend | |
Dyspneu | Niet bekend | |
Hypoxie | Niet bekend | |
Hypotensie | Niet bekend | |
Angio‑oedeem | Niet bekend | |
Zenuwstelselaandoeningen | Brandend gevoel | Vaak |
Bloedvataandoeningen | Flushing | Zeer vaak |
Opvlieger | Vaak | |
Ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen | Rhinorroe | Niet bekend |
Maagdarmstelselaandoeningen | Diarree | Zeer vaak |
Nausea | Zeer vaak | |
Pijn in de bovenbuik | Zeer vaak | |
Buikpijn | Zeer vaak | |
Braken | Vaak | |
Dyspepsie | Vaak | |
Gastritis | Vaak | |
Maag‑darmstoornis | Vaak | |
Lever‑ en galaandoeningen | Aspartaataminotransferase verhoogd | Vaak |
Alanineaminotransferase verhoogd | Vaak | |
Geneesmiddelgeïnduceerd leverletsel | Zelden | |
Huid‑ en onderhuidaandoeningen | Pruritus | Vaak |
Rash | Vaak | |
Erytheem | Vaak | |
Alopecia | Vaak | |
Nier‑ en urinewegaandoeningen | Proteïnurie | Vaak |
Algemene aandoeningen en toedieningsplaatsstoornissen | Warm aanvoelen | Vaak |
Onderzoeken | Ketonen in de urine | Zeer vaak |
Albumine aanwezig in de urine | Vaak | |
Aantal witte bloedcellen verlaagd | Vaak |
Beschrijving van geselecteerde bijwerkingen
Flushing
In de placebogecontroleerde onderzoeken was de incidentie van flushing (34% versus 4%) en opvliegers (7% versus 2%) hoger bij patiënten die met Tecfidera werden behandeld vergeleken bij die met placebo waren behandeld. Flushing wordt meestal beschreven als roodheid of opvliegers, maar kan ook warmte, roodheid, jeuk en brandend gevoel omvatten. Flushing komt vaak aan het begin van de behandeling voor (voornamelijk gedurende de eerste maand) en bij patiënten die flushing ondervinden kan dit tijdens de gehele behandeling met Tecfidera met tussenpozen blijven optreden. Bij patiënten met flushing had de meerderheid flushingverschijnselen die qua ernst licht of matig waren. In totaal is 3% van de met Tecfidera behandelde patiënten gestopt met de behandeling wegens flushing. De incidentie van ernstige flushing, die kan worden gekarakteriseerd door gegeneraliseerd erytheem, uitslag en/of pruritus, werd waargenomen bij minder dan 1% van de met Tecfidera behandelde patiënten (zie rubriek 4.2, 4.4 en 4.5).
Maag‑darmbijwerkingen
De incidentie van maag‑darmbijwerkingen (bijv. diarree [14% versus 10%], misselijkheid [12% versus 9%], pijn in de bovenbuik [10% versus 6%], buikpijn [9% versus 4%], braken [8% versus 5%] en dyspepsie [5% versus 3%]) was hoger bij patiënten behandeld met Tecfidera vergeleken met patiënten behandeld met placebo. Maag‑darmbijwerkingen komen vaak aan het begin van de behandeling voor (voornamelijk gedurende de eerste maand) en bij patiënten die maag‑darmbijwerkingen ondervinden kan dit gedurende de gehele behandeling met Tecfidera met tussenpozen blijven optreden. Bij de meeste patiënten die maag‑darmbijwerkingen ervoeren, waren deze licht tot matig van ernst. Vier procent (4%) van met Tecfidera behandelde patiënten heeft de behandeling stopgezet wegens maag‑darmbijwerkingen. De incidentie van ernstige maag‑darmbijwerkingen, waaronder gastro‑enteritis en gastritis, werd waargenomen bij 1% van de met Tecfidera behandelde patiënten (zie rubriek 4.2).
Leverfunctie
Op basis van gegevens van placebogecontroleerde onderzoeken had de meerderheid van patiënten met een stijging levertransaminasen die < 3 keer zo hoog waren als de ULN. De toegenomen incidentie van stijgingen van levertransaminasen bij de met Tecfidera behandelde patiënten vergeleken met placebo werd voornamelijk waargenomen gedurende de eerste 6 maanden van behandeling. Stijgingen in respectievelijk alanineaminotransferase en aspartaataminotransferase ≥ 3 keer ULN werden waargenomen bij 5% en 2% van de met placebo behandelde patiënten en bij 6% en 2% van de met Tecfidera behandelde patiënten. Stopzetting wegens verhoogde levertransaminasen bedroeg < 1% en was vergelijkbaar bij patiënten behandeld met Tecfidera of placebo. Stijgingen in transaminasen ≥ 3 keer ULN met gelijktijdige stijgingen in totaalbilirubine > 2 keer ULN zijn niet waargenomen in placebogecontroleerde onderzoeken.
Stijging van leverenzymwaarden en gevallen van geneesmiddelgeïnduceerd leverletsel (stijgingen in transaminasen ≥ 3 keer ULN met gelijktijdige stijgingen in totaalbilirubine > 2 keer ULN) zijn gemeld in postmarketingervaring na toediening van Tecfidera, die verdwenen na stopzetting van de behandeling.
Lymfopenie
In de placebogecontroleerde studies hadden de meeste patiënten (> 98%) normale lymfocytenaantallen voordat de behandeling werd geïnitieerd. Bij behandeling met Tecfidera nam het gemiddelde aantal lymfocyten gedurende het eerste jaar af en bleef daarna stabiel. Gemiddeld nam het aantal lymfocyten met ongeveer 30% af t.o.v. de uitgangssituatie. Het gemiddelde en mediane aantal lymfocyten bleef binnen de normale limieten. Lymfocytenaantallen < 0,5 × 109/l werden waargenomen bij < 1% van patiënten behandeld met placebo en 6% van patiënten behandeld met Tecfidera. Een lymfocytenaantal < 0,2 × 109/l werd waargenomen bij 1 patiënt behandeld met Tecfidera en bij geen patiënten behandeld met placebo.
In klinische onderzoeken (zowel gecontroleerde als ongecontroleerde) had 41% van de patiënten die werd behandeld met Tecfidera lymfopenie (in deze onderzoeken gedefinieerd als < 0,91 × 109/l). Milde lymfopenie (aantallen van ≥ 0,8 × 109/l tot < 0,91 × 109/l) werd waargenomen bij 28% van de patiënten; matige lymfopenie (aantallen ≥ 0,5 × 109/l tot < 0,8 × 109/l) die gedurende minimaal zes maanden aanhield werd waargenomen bij 11% van de patiënten; ernstige lymfopenie (aantallen < 0,5 × 109/l) die gedurende minimaal zes maanden aanhield werd waargenomen bij 2% van de patiënten. In de groep met ernstige lymfopenie bleef het overgrote deel van de lymfocytenaantallen < 0,5 × 109/l bij voortzetting van de behandeling.
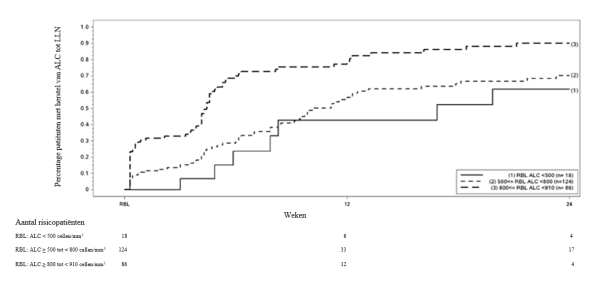
Daarnaast is gebleken dat in een ongecontroleerde, prospectief, postmarketingonderzoek in week 48 van de behandeling met Tecfidera week 48 van de behandeling met Tecfidera (n=185) CD4+‑T‑cellen matig (aantallen ≥ 0,2 × 109/l tot < 0,4 × 109/l) of sterk (< 0,2 × 109/l) waren verminderd bij maximaal 37% respectievelijk 6% van de patiënten, terwijl CD8+‑T‑cellen vaker verminderd waren bij maximaal 59% van de patiënten bij aantallen < 0,2 × 109/l en 25% van de patiënten bij aantallen < 0,1 × 109/l. In gecontroleerde en ongecontroleerde klinische onderzoeken werden patiënten die de behandeling met Tecfidera stopzetten en die een lymfocytenaantal lager dan de LLN hadden, gevolgd tot aan herstel van het lymfocytenaantal tot de LLN (zie rubriek 5.1).
Progressieve multifocale leuko‑encefalopathie (PML)
Bij behandeling met Tecfidera zijn gevallen gemeld van infecties met John Cunningham-virus (JCV) die PML veroorzaakte (zie rubriek 4.4). PML kan fataal aflopen of leiden tot ernstige invaliditeit. In een van de klinische onderzoeken heeft 1 patiënt die Tecfidera innam PML ontwikkeld in combinatie met langdurige ernstige lymfopenie (lymfocytenaantallen van < 0,5 × 109/l gedurende 3,5 jaar) met fatale afloop. In de postmarketingsetting is ook PML opgetreden in de aanwezigheid van matige en milde lymfopenie (> 0,5 × 109/l tot < LLN zoals gedefinieerd door het referentiebereik van plaatselijke laboratoria).
In meerdere PML‑gevallen waarbij de subreeksen van T‑cellen zijn vastgesteld op het moment dat PML is vastgesteld, bleken CD8+‑T‑celaantallen tot < 0,1 × 109/l te zijn afgenomen, terwijl verminderingen in CD4+‑T‑celaantallen variabel waren (variërend van < 0,05 tot 0,5 × 109/l) en sterker correleerden met de algemene ernst van lymfopenie (< 0,5 × 109/l tot < LLN). Als resultaat was de CD4+/CD8+‑verhouding bij deze patiënten verhoogd.
Langdurige matige tot ernstige lymfopenie lijkt het risico op PML bij gebruik van Tecfidera te vergroten. Bij patiënten met milde lymfopenie is echter ook PML opgetreden. Bovendien zijn de meeste gevallen van PML in de postmarketingsetting opgetreden bij patiënten > 50 jaar.
Herpes zoster‑infecties
Herpes zoster-infecties zijn gemeld met Tecfidera. In het doorlopende langdurige extensieonderzoek waarin 1.736 MS‑patiënten werden behandeld, kwam bij ongeveer 5% van de proefpersonen een of meerdere infecties met herpes zoster voor, waarvan 42% licht, 55% matig en 3% ernstig van aard waren. De tijd vanaf de eerste dosis Tecfidera tot het eerste optreden varieerde van ongeveer 3 maanden tot 10 jaar. Vier patiënten hadden ernstige voorvallen die allemaal verdwenen. Bij de meeste proefpersonen, waaronder de proefpersonen die een ernstige infectie met herpes zoster hadden, lagen de lymfocytenaantallen hoger dan de ondergrens van normaal. Bij de meeste proefpersonen met gelijktijdig lymfocytenaantallen lager dan de LLN werd de lymfopenie geclassificeerd als matig of ernstig. In de postmarketingsetting waren de meeste gevallen van infectie met herpes zoster niet ernstig en genezen na behandeling. Er zijn beperkte gegevens beschikbaar over de absolute lymfocytenaantallen (absolute lymphocyte count, ALC) bij patiënten met infectie met herpes zoster in de postmarketingsetting. Bij melding hadden de meeste patiënten echter matige (≥ 0,5 × 109/l tot < 0,8 × 109/l) of ernstige (< 0,5 × 109/l tot 0,2 × 109/l) lymfopenie (zie rubriek 4.4).
Laboratoriumafwijkingen
In de placebogecontroleerde studies werden meer urineketonen (1+ of groter) gemeten bij patiënten behandeld met Tecfidera (45%) vergeleken met die behandeld met placebo (10%). In klinische studies werden geen ongewenste klinische gevolgen waargenomen.
1,25‑dihydroxyvitamine D‑gehaltes namen af bij de met Tecfidera behandelde patiënten vergeleken met placebo (mediaan percentage afname t.o.v. de uitgangswaarde na 2 jaar was 25% versus 15%) en parathyroïdhormoon (PTH)‑gehaltes namen toe bij de met Tecfidera behandelde patiënten vergeleken met placebo (mediaan percentage toename t.o.v. de uitgangswaarde na 2 jaar was 29% versus 15%). De gemiddelde waarden voor beide parameters bleven binnen het normale bereik.
Een tijdelijke toename in het gemiddelde aantal eosinofielen werd gedurende de eerste 2 maanden van de behandeling waargenomen.
Pediatrische patiënten
In een 96 weken durend, open‑label, gerandomiseerd onderzoek met werkzame controle werden pediatrische patiënten met RRMS (n=7 in de leeftijd van 10 tot en met 12 jaar en n=71 in de leeftijd van 13 tot en met 17 jaar) behandeld met 120 mg tweemaal per dag gedurende 7 dagen, gevolgd door 240 mg tweemaal per dag gedurende de rest van de behandeling. Het veiligheidsprofiel bleek bij pediatrische patiënten vergelijkbaar met het profiel dat eerder werd waargenomen bij volwassen patiënten.
De opzet van het pediatrische klinische onderzoek verschilde van die bij de placebogecontroleerde klinische onderzoeken bij volwassenen. Daarom kan niet worden uitgesloten dat de opzet van de klinische onderzoeken heeft bijgedragen aan de numerieke verschillen in bijwerkingen tussen de pediatrische en de volwassen populatie. Maagdarmstelselaandoeningen alsook ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen en de bijwerkingen hoofdpijn en dysmenorroe werden frequenter gemeld (≥ 10%) bij pediatrische patiënten dan bij volwassen patiënten.
Deze bijwerkingen werden met de volgende percentages gemeld bij pediatrische patiënten:
- Hoofdpijn werd gemeld bij 28% van de met Tecfidera behandelde patiënten versus 36% van de patiënten behandeld met interferon bèta‑1a.
- Maagdarmstelselaandoeningen werden gemeld bij 74% van de met Tecfidera behandelde patiënten versus 31% van de patiënten behandeld met interferon bèta‑1a. Daarvan werden buikpijn en braken het frequentst gemeld bij het gebruik van Tecfidera.
- Ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen werden gemeld bij 32% van de met Tecfidera behandelde patiënten versus 11% van de patiënten behandeld met interferon bèta‑1a. Daarvan werden orofaryngeale pijn en hoesten het frequentst gemeld bij het gebruik van Tecfidera.
- Dysmenorroe werd gemeld bij 17% van de met Tecfidera behandelde patiënten versus 7% van de patiënten behandeld met interferon bèta‑1a.
In een klein, 24 weken durend, open‑label ongecontroleerd onderzoek bij pediatrische patiënten met RRMS in de leeftijd van 13 tot en met 17 jaar (120 mg tweemaal per dag gedurende 7 dagen, gevolgd door 240 mg tweemaal per dag gedurende de rest van de behandeling; n=22), gevolgd door een 96 weken durend extensieonderzoek (240 mg tweemaal per dag; n=20) bleek het veiligheidsprofiel vergelijkbaar met het profiel dat werd waargenomen bij volwassen patiënten.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
Nederland
Nederlands Bijwerkingen Centrum Lareb
Website: www.lareb.nl
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Biogen Netherlands B.V.
Prins Mauritslaan 13
1171 LP Badhoevedorp
Nederland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/13/837/001
EU/1/13/837/002
EU/1/13/837/003
10. DATUM VAN HERZIENING VAN DE TEKST
12/2024
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3236080 | TECFIDERA 120MG MAAGSAPRESIST CAPS 14 | L04AX07 | € 98,73 | - | Ja | € 12,8 | € 8,5 |
| 3236106 | TECFIDERA 240MG MAAGSAPRESIST CAPS 56 | L04AX07 | € 362 | - | Ja | € 12,8 | € 8,5 |