1. NAAM VAN HET GENEESMIDDEL
XELJANZ 5 mg filmomhulde tabletten
XELJANZ 10 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
XELJANZ 5 mg filmomhulde tabletten
Elke filmomhulde tablet bevat tofacitinibcitraat, overeenkomend met 5 mg tofacitinib.
Hulpstof met bekend effect
Elke filmomhulde tablet bevat 59,44 mg lactose.
XELJANZ 10 mg filmomhulde tabletten
Elke filmomhulde tablet bevat tofacitinibcitraat, overeenkomend met 10 mg tofacitinib.
Hulpstof met bekend effect
Elke filmomhulde tablet bevat 118,88 mg lactose.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet)
XELJANZ 5 mg filmomhulde tabletten
Witte, ronde tablet met een diameter van 7,9 mm, met aan de ene zijde de inscriptie 'Pfizer' en aan de andere zijde 'JKI 5'.
XELJANZ 10 mg filmomhulde tabletten
Blauwe, ronde tablet met een diameter van 9,5 mm, met aan de ene zijde de inscriptie 'Pfizer' en aan de andere zijde 'JKI 10'.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Reumatoïde artritis
Tofacitinib in combinatie met methotrexaat (MTX) is geïndiceerd voor de behandeling van matige tot ernstige actieve reumatoïde artritis (RA) bij volwassen patiënten die onvoldoende reageerden op of intolerant zijn voor één of meerdere disease-modifying anti-rheumatic drugs (DMARD’s) (zie rubriek 5.1). Tofacitinib kan worden gegeven als monotherapie indien MTX niet wordt verdragen of indien behandeling met MTX niet gepast is (zie rubriek 4.4 en 4.5).
Arthritis psoriatica
Tofacitinib in combinatie met MTX is geïndiceerd voor de behandeling van actieve arthritis psoriatica (PsA) bij volwassen patiënten die onvoldoende hebben gereageerd op of intolerant waren voor een eerdere behandeling met een disease‑modifying anti-rheumatic drug (DMARD) (zie rubriek 5.1).
Spondylitis ankylopoetica
Tofacitinib is geïndiceerd voor de behandeling van actieve spondylitis ankylopoetica (SA) bij volwassen patiënten die onvoldoende reageerden op conventionele behandeling.
Colitis ulcerosa
Tofacitinib is geïndiceerd voor de behandeling van matig tot ernstig actieve colitis ulcerosa (Ulcerative Colitis [UC]) bij volwassen patiënten die onvoldoende reageerden op, niet meer reageerden op of intolerant waren voor ofwel conventionele behandeling ofwel voor een biologisch middel (zie rubriek 5.1).
Juveniele idiopathische artritis (JIA)
Tofacitinib is geïndiceerd voor de behandeling van actieve polyarticulaire juveniele idiopathische artritis (reumafactor-positieve [RF+] of ‑negatieve [RF-] polyartritis en uitgebreide oligoartritis) en juveniele artritis psoriatica (PsA) bij patiënten van 2 jaar en ouder, die onvoldoende hebben gereageerd op eerdere behandeling met DMARD’s.
Tofacitinib kan worden gegeven in combinatie met methotrexaat (MTX) of als monotherapie indien MTX niet wordt verdragen of indien verdere behandeling met MTX niet gepast is.
4.2 Dosering en wijze van toediening
De behandeling dient te worden gestart en plaats te vinden onder toezicht van gespecialiseerde artsen met ervaring in de diagnosticering en behandeling van aandoeningen waarvoor tofacitinib is geïndiceerd.
Dosering
Reumatoïde artritis en arthritis psoriatica
De aanbevolen dosis is 5 mg filmomhulde tabletten, tweemaal daags toegediend. Deze dosis dient niet te worden overschreden.
Er is geen dosisaanpassing nodig bij gebruik in combinatie met MTX.
Zie tabel 1 voor informatie over het wisselen tussen tofacitinib filmomhulde tabletten en tofacitinib tabletten met verlengde afgifte.
Tabel 1: Wisselen tussen tofacitinib filmomhulde tabletten en tofacitinib tabletten met verlengde afgifte
Wisselen tussen tofacitinib 5 mg filmomhulde tabletten en tofacitinib 11 mg tablet met verlengde afgiftea | Er kan onderling worden gewisseld tussen behandeling met tweemaal daags tofacitinib 5 mg filmomhulde tabletten en eenmaal daags tofacitinib 11 mg tablet met verlengde afgifte op de dag na de laatste dosis van een van beide tabletten. |
a Zie rubriek 5.2 voor een vergelijking van de farmacokinetiek van formuleringen met verlengde afgifte en die van filmomhulde formuleringen. | |
Spondylitis ankylopoetica
De aanbevolen dosis tofacitinib is 5 mg, tweemaal daags toegediend.
Colitis ulcerosa
Inductiebehandeling
De aanbevolen dosis is 10 mg, tweemaal daags oraal toegediend als inductie gedurende 8 weken.
Voor patiënten die onvoldoende therapeutisch voordeel bereiken na week 8 kan de inductiedosis van tweemaal daags 10 mg met 8 weken worden verlengd (16 weken in totaal), gevolgd door tweemaal daags 5 mg als onderhoud. De inductiebehandeling met tofacitinib dient te worden stopgezet bij iedere patiënt die na week 16 geen aanwijzingen voor therapeutisch voordeel vertoont.
Onderhoudsbehandeling
De aanbevolen dosis voor onderhoudsbehandeling is tweemaal daags 5 mg tofacitinib oraal toegediend.
Tofacitinib tweemaal daags 10 mg als onderhoudsbehandeling wordt niet aanbevolen bij patiënten met UC die bekende risicofactoren hebben voor veneuze trombo-embolie (VTE), ernstige ongewenste cardiovasculaire voorvallen (MACE, major adverse cardiovascular events) en maligniteiten, tenzij er geen geschikte alternatieve behandeling is (zie rubriek 4.4 en 4.8).
Voor patiënten met UC die geen verhoogd risico op VTE, MACE en maligniteiten hebben (zie rubriek 4.4) kan tweemaal daags 10 mg tofacitinib oraal toegediend, worden overwogen als de respons van de patiënt op tweemaal daags 5 mg tofacitinib onvoldoende is en de patiënt niet reageerde op alternatieve behandelopties voor colitis ulcerosa, zoals behandeling met een tumornecrosefactorremmer (TNF‑remmer).
Tofacitinib tweemaal daags 10 mg als onderhoudsbehandeling dient zo kort mogelijk te worden gebruikt. De laagste effectieve dosis die nodig is voor een aanhoudende respons moet worden gebruikt.
Bij patiënten die reageren op behandeling met tofacitinib, kan het gebruik van corticosteroïden worden verminderd en/of stopgezet in overeenstemming met de standaardzorg.
Herbehandeling bij UC
Indien de behandeling werd onderbroken, kan worden overwogen de behandeling met tofacitinib opnieuw te starten. Als de respons is verdwenen, kan herinductie met tweemaal daags 10 mg tofacitinib worden overwogen. De periode van onderbreking van de behandeling in klinische onderzoeken duurde maximaal 1 jaar. De werkzaamheid kan opnieuw behaald worden na 8 weken behandeling met tweemaal daags 10 mg (zie rubriek 5.1).
Polyarticulaire JIA en juveniele PsA (kinderen van 2 tot 18 jaar)
Tofacitinib kan worden gebruikt als monotherapie of in combinatie met MTX.
De aanbevolen dosering bij patiënten van 2 jaar en ouder is gebaseerd op de volgende gewichtscategorieën:
Tabel 2: Dosis tofacitinib voor patiënten van twee jaar en ouder met polyarticulaire juveniele idiopathische artritis en juveniele PsA
Lichaamsgewicht (kg) | Dosering |
10‑<20 | 3,2 mg (3,2 ml drank) tweemaal daags |
20‑<40 | 4 mg (4 ml drank) tweemaal daags |
≥40 | 5 mg (5 ml drank of 5 mg filmomhulde tablet) tweemaal daags |
Patiënten van 40 kg die worden behandeld met tweemaal daags 5 ml tofacitinib drank kunnen wisselen naar tweemaal daags tofacitinib 5 mg filmomhulde tabletten. Patiënten < 40 kg kunnen niet wisselen vanaf tofacitinib drank.
Onderbreking en stopzetting van de toediening bij volwassenen en pediatrische patiënten
Indien een patiënt een ernstige infectie ontwikkelt, dient de behandeling met tofacitinib te worden onderbroken totdat de infectie onder controle is.
Onderbreking van de toediening kan nodig zijn voor de behandeling van aan de dosis gerelateerde laboratoriumafwijkingen, waaronder lymfopenie, neutropenie en anemie. Zoals beschreven in de tabellen 3, 4 en 5 hieronder worden aanbevelingen voor de tijdelijke onderbreking of het definitief stoppen van de behandeling gedaan op basis van de ernst van de laboratoriumafwijkingen (zie rubriek 4.4).
Starten van de toediening wordt niet aanbevolen bij patiënten met een absoluut lymfocytenaantal (ALC) lager dan 750 cellen/mm3.
Tabel 3: Laag absoluut lymfocytenaantal
Laag absoluut lymfocytenaantal (ALC) (zie rubriek 4.4) | |
Laboratoriumwaarde | Aanbeveling |
ALC hoger dan of gelijk aan 750 | De dosis dient te worden gehandhaafd. |
ALC 500–750 | Bij aanhoudende dalingen tussen deze waarden (twee opeenvolgende routinetesten tussen deze waarden) dient de toediening te worden verlaagd of onderbroken. |
ALC lager dan 500 | Indien de laboratoriumwaarde wordt bevestigd door herhaald testen binnen 7 dagen dient de toediening te worden stopgezet. |
Starten van de toediening wordt niet aanbevolen bij volwassen patiënten met een absoluut neutrofielenaantal (ANC) lager dan 1.000 cellen/mm3. Starten van de toediening wordt niet aanbevolen bij pediatrische patiënten met een absoluut neutrofielenaantal (ANC) lager dan 1.200 cellen/mm3.
Tabel 4: Laag absoluut neutrofielenaantal
Laag absoluut neutrofielenaantal (ANC) (zie rubriek 4.4) | |
Laboratoriumwaarde | Aanbeveling |
ANC hoger dan 1.000 | De dosis dient te worden gehandhaafd. |
ANC 500–1.000 | Bij aanhoudende dalingen tussen deze waarden (twee opeenvolgende routinetesten tussen deze waarden) dient de toediening te worden verlaagd of onderbroken. |
ANC lager dan 500 | Indien de laboratoriumwaarde wordt bevestigd door herhaald testen binnen 7 dagen dient de toediening te worden stopgezet. |
Starten van de toediening wordt niet aanbevolen bij volwassen patiënten met een hemoglobinewaarde lager dan 5,6 mmol/l (9 g/dl). Starten van de toediening wordt niet aanbevolen bij pediatrische patiënten met een hemoglobinewaarde lager dan 6,1 mmol/l (10 g/dl).
Tabel 5: Lage hemoglobinewaarde
Lage hemoglobinewaarde (zie rubriek 4.4) | |
Laboratoriumwaarde | Aanbeveling |
Minder dan of gelijk aan 1,24 mmol/l (2 g/dl) daling en hoger dan of gelijk aan 5,6 mmol/l (9,0 g/dl) | De dosis dient te worden gehandhaafd. |
Meer dan 1,24 mmol/l (2 g/dl) daling of lager dan 5,0 mmol/l (8,0 g/dl) | De toediening dient te worden onderbroken totdat de hemoglobinewaarden zijn genormaliseerd. |
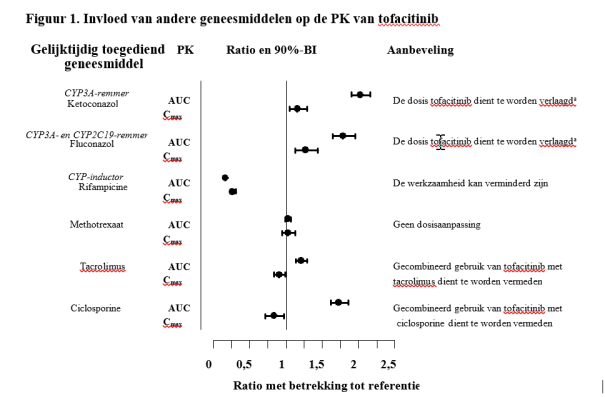
Interacties
De totale dagelijkse dosis tofacitinib dient te worden gehalveerd bij patiënten die krachtige remmers van cytochroom (CYP) P450 3A4 (bijv. ketoconazol) krijgen en bij patiënten die gelijktijdig 1 of meer geneesmiddelen krijgen die leiden tot zowel een matige remming van CYP3A4 als een krachtige remming van CYP2C19 (bijv. fluconazol) (zie rubriek 4.5) als volgt:
- De dosis tofacitinib dient te worden verlaagd naar eenmaal daags 5 mg bij patiënten die tweemaal daags 5 mg krijgen (volwassen en pediatrische patiënten).
- De dosis tofacitinib dient te worden verlaagd naar tweemaal daags 5 mg bij patiënten die tweemaal daags 10 mg krijgen (volwassen patiënten).
Alleen bij pediatrische patiënten: beschikbare gegevens duiden erop dat binnen 18 weken na aanvang van de behandeling met tofacitinib klinische verbetering wordt waargenomen. Bij een patiënt die binnen dit tijdsbestek geen klinische verbetering laat zien, dient het voortzetten van de behandeling zorgvuldig te worden heroverwogen.
Stopzetting van toediening bij SA
Beschikbare gegevens duiden erop dat binnen 16 weken na aanvang van de behandeling met tofacitinib klinische verbetering van SA wordt waargenomen. Bij een patiënt die binnen dit tijdsbestek geen klinische verbetering laat zien, dient het voortzetten van de behandeling zorgvuldig te worden heroverwogen.
Bijzondere populaties
Ouderen
Er is geen dosisaanpassing nodig bij patiënten van 65 jaar en ouder. Er zijn beperkte gegevens beschikbaar bij patiënten van 75 jaar en ouder. Zie rubriek 4.4 voor Gebruik bij patiënten van 65 jaar en ouder.
Leverinsufficiëntie
Tabel 6: Dosisaanpassing voor leverinsufficiëntie
Categorie leverinsufficiëntie | Classificatie | Dosisaanpassing bij leverinsufficiëntie voor tabletten met verschillende sterkte |
Licht | Child Pugh A | Geen dosisaanpassing nodig. |
Matig | Child Pugh B | De dosis dient te worden verlaagd naar eenmaal daags 5 mg, wanneer de aangegeven dosis bij een normale leverfunctie tweemaal daags 5 mg is. |
Ernstig | Child Pugh C | Tofacitinib dient niet te worden gebruikt bij patiënten met ernstige leverinsufficiëntie (zie rubriek 4.3). |
Nierinsufficiëntie
Tabel 7: Dosisaanpassing voor nierinsufficiëntie
Categorie nierinsufficiëntie | Creatinineklaring | Dosisaanpassing bij nierinsufficiëntie voor tabletten met verschillende sterkte |
Licht | 50-80 ml/min | Geen dosisaanpassing nodig. |
Matig | 30-49 ml/min | Geen dosisaanpassing nodig. |
Ernstig (inclusief patiënten die hemodialyse ondergaan) | < 30 ml/min | De dosis dient te worden verlaagd naar eenmaal daags 5 mg, wanneer de aangegeven dosis bij een normale nierfunctie tweemaal daags 5 mg is. |
Pediatrische patiënten
De veiligheid en werkzaamheid van tofacitinib bij kinderen jonger dan 2 jaar met polyarticulaire JIA en juveniele PsA zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
De veiligheid en werkzaamheid van tofacitinib bij kinderen jonger dan 18 jaar met andere indicaties (bijv. colitis ulcerosa) zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Oraal gebruik.
Tofacitinib wordt oraal gegeven, met of zonder voedsel.
Voor patiënten die moeite hebben met slikken, mogen de tofacitinib tabletten worden geplet en met water worden ingenomen.
4.3 Contra-indicaties
- Overgevoeligheid voor de werkzame stof(fen) of voor een van de in rubriek 6.1 vermelde hulpstof(fen).
- Actieve tuberculose (tbc), ernstige infecties zoals sepsis, of opportunistische infecties (zie rubriek 4.4).
- Ernstige leverinsufficiëntie (zie rubriek 4.2).
- Zwangerschap en borstvoeding (zie rubriek 4.6).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Reumatoïde artritis
De vaakst voorkomende ernstige bijwerkingen waren ernstige infecties (zie rubriek 4.4). In de langetermijnveiligheidspopulatie met alle blootstellingen waren de vaakst voorkomende ernstige infecties die zijn gemeld met tofacitinib pneumonie (1,7%), herpes zoster (0,6%), urineweginfectie (0,4%), cellulitis (0,4%), diverticulitis (0,3%) en appendicitis (0,2%). Als opportunistische infecties werden tbc en andere mycobacteriële infecties, cryptokokken, histoplasmose, oesofageale candidiasis, multidermatomale herpes zoster, cytomegalovirusinfectie, BK-virusinfecties en listeriosis gemeld met tofacitinib. Sommige patiënten hadden een verspreide ziekte in plaats van een gelokaliseerde ziekte. Er kunnen ook andere ernstige infecties optreden die niet in klinische onderzoeken werden gemeld (bijv. coccidioïdomycose).
De vaakst gemelde bijwerkingen tijdens de eerste 3 maanden van de dubbelblinde, placebo- of MTX- gecontroleerde klinische onderzoeken waren hoofdpijn (3,9%), bovensteluchtweginfecties (3,8%), virale bovensteluchtweginfectie (3,3%), diarree (2,9%), nausea (2,7%) en hypertensie (2,2%).
Het percentage patiënten dat stopte met de behandeling vanwege bijwerkingen tijdens de eerste 3 maanden van de dubbelblinde, placebo- of methotrexaat-gecontroleerde onderzoeken bedroeg 3,8% voor patiënten die tofacitinib namen. De vaakst voorkomende infecties die leidden tot stopzetting van de behandeling gedurende de eerste 3 maanden in gecontroleerde klinische onderzoeken waren herpes zoster (0,19%) en pneumonie (0,15%).
Arthritis psoriatica
Het veiligheidsprofiel dat werd waargenomen bij patiënten met actieve PsA die werden behandeld met tofacitinib, kwam over het algemeen overeen met het veiligheidsprofiel dat werd waargenomen bij patiënten met RA die werden behandeld met tofacitinib.
Spondylitis ankylopoetica
Het veiligheidsprofiel dat werd waargenomen bij patiënten met actieve SA die werden behandeld met tofacitinib, kwam over het algemeen overeen met het veiligheidsprofiel dat werd waargenomen bij patiënten met RA die werden behandeld met tofacitinib.
Colitis ulcerosa
De vaakst gemelde bijwerkingen bij patiënten die tweemaal daags 10 mg tofacitinib kregen in de inductieonderzoeken, waren hoofdpijn, nasofaryngitis, nausea en artralgie.
In de inductie- en onderhoudsonderzoeken waren de vaakst voorkomende categorieën ernstige bijwerkingen in de tofacitinib- en placebobehandelgroepen gastro-intestinale aandoeningen en infecties en de vaakst voorkomende ernstige bijwerking was verergering van UC.
Over het algemeen kwam het veiligheidsprofiel dat werd waargenomen bij patiënten met UC die werden behandeld met tofacitinib, overeen met het veiligheidsprofiel van tofacitinib voor de indicatie RA.
Overzichtstabel van bijwerkingen
De bijwerkingen die worden vermeld in onderstaande tabel zijn afkomstig van klinische onderzoeken bij patiënten met RA, PsA, SA en UC en worden weergegeven per systeem/orgaanklasse (SOC) en frequentiecategorie, gedefinieerd met de volgende conventie: zeer vaak (≥1/10), vaak (≥1/100, <1/10), soms (≥1/1.000, <1/100), zelden (≥1/10.000, <1/1.000), zeer zelden (<1/10.000) of niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiecategorie worden de bijwerkingen weergegeven in volgorde van afnemende ernst.
Tabel 8: Bijwerkingen
Systeem/ | Vaak | Soms | Zelden | Zeer zelden | Niet bekend (kan met de beschikbare gegevens niet worden bepaald) |
Infecties en parasitaire aandoeningen | Pneumonie | Tuberculose | Sepsis | Tuberculose van het centrale zenuwstelsel |
|
Neoplasmata, benigne, maligne en niet-gespecificeerd (inclusief cysten en poliepen) |
| Longkanker | Lymfoom |
|
|
Bloed- en lymfestelselaandoeningen | Lymfopenie | Leukopenie |
|
|
|
Immuunsysteem-aandoeningen |
|
|
|
| Overgevoeligheid* |
Voedings- en stofwisselingsstoornissen |
| Dyslipidemie |
|
|
|
Psychische stoornissen |
| Insomnia |
|
|
|
Zenuwstelselaandoeningen | Hoofdpijn | Paresthesie |
|
|
|
Hartaandoeningen |
| Myocardinfarct |
|
|
|
Bloedvataandoeningen | Hypertensie | Veneuze trombo-embolie** |
|
|
|
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen | Hoesten | Dyspneu |
|
|
|
Maagdarmstelselaandoeningen | Buikpijn |
|
|
|
|
Lever- en galaandoeningen |
| Hepatische steatose | Leverfunctietesten abnormaal |
|
|
Huid- en onderhuidaandoeningen | Rash Acne | Erytheem |
|
|
|
Skeletspierstelsel- en bindweefselaandoeningen | Artralgie | Gewrichtszwelling | Skeletspierstelselpijn |
|
|
Algemene aandoeningen en toedieningsplaatsstoornissen | Perifeer oedeem | Pyrexie |
|
|
|
Onderzoeken | Creatinefosfokinase in bloed verhoogd | Creatinine in bloed verhoogd |
|
|
|
Letsels, intoxicaties en verrichtingscomplicaties |
| Ligamentverstuiking |
|
|
|
*Data uit spontane rapportage
**Onder veneuze trombo-embolie vallen PE, DVT, retinale veneuze trombose en cerebrale veneuze sinus trombose
Beschrijving van geselecteerde bijwerkingen
Veneuze trombo-embolie
Reumatoïde artritis
In een groot (N=4.362), gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met reumatoïde artritis van 50 jaar en ouder die ten minste één bijkomende cardiovasculaire (CV) risicofactor hadden, werd VTE waargenomen met een verhoogde en dosisafhankelijke incidentie bij patiënten behandeld met tofacitinib ten opzichte van patiënten behandeld met TNF‑remmers (zie rubriek 5.1). De meeste van deze voorvallen waren ernstig en sommige leidden tot de dood. De incidentiecijfers (95%‑BI) voor PE voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib, en TNF‑remmers bedroegen respectievelijk 0,17 (0,08; 0,33), 0,50 (0,32; 0,74) en 0,06 (0,01; 0,17) patiënten met voorvallen per 100 patiëntjaren. Vergeleken met TNF‑remmers was de hazardratio (HR) voor PE 2,93 (0,79; 10,83) en 8,26 (2,49; 27,43) voor respectievelijk tweemaal daags 5 mg tofacitinib en tweemaal daags 10 mg tofacitinib (zie rubriek 5.1). Van de met tofacitinib behandelde patiënten bij wie PE werd waargenomen, had de meerderheid (97%) risicofactoren voor VTE.
Spondylitis ankylopoetica
In de gecombineerde, gerandomiseerde, gecontroleerde klinische fase 2- en fase 3-onderzoeken waren er geen gevallen van VTE bij 420 patiënten (233 patiëntjaren aan observatie) die maximaal 48 weken tofacitinib kregen.
Colitis ulcerosa (UC)
In het lopende lange termijn UC-onderzoek zijn gevallen van PE en DVT waargenomen bij patiënten met onderliggende risicofactoren voor VTE die tweemaal daags 10 mg tofacitinib gebruiken.
Alle infecties
Reumatoïde artritis
In gecontroleerde klinische fase 3-onderzoeken bedroegen de percentages voor infecties over een periode van 0 tot 3 maanden in de groep met tweemaal daags 5 mg tofacitinib als monotherapie (totaal 616 patiënten) en de groep met tweemaal daags 10 mg tofacitinib als monotherapie (totaal 642 patiënten) respectievelijk 16,2% (100 patiënten) en 17,9% (115 patiënten), vergeleken met 18,9% (23 patiënten) in de placebogroep (totaal 122 patiënten). In gecontroleerde klinische fase 3-onderzoeken met achtergrond-DMARD's bedroegen de percentages voor infecties over een periode van 0 tot 3 maanden in de groep met tweemaal daags 5 mg tofacitinib plus DMARD (totaal 973 patiënten) en in de groep met tweemaal daags 10 mg tofacitinib plus DMARD (totaal 969 patiënten) respectievelijk 21,3% (207 patiënten) en 21,8% (211 patiënten), vergeleken met 18,4% (103 patiënten) in de groep met placebo plus DMARD (totaal 559 patiënten).
De vaakst gemelde infecties waren bovensteluchtweginfecties en nasofaryngitis (respectievelijk 3,7% en 3,2%).
Het totale incidentiecijfer voor infecties met tofacitinib in de langetermijnveiligheidspopulatie met alle blootstellingen (totaal 4.867 patiënten) bedroeg 46,1 patiënten met voorvallen per 100 patiëntjaren (43,8 en 47,2 patiënten met voorvallen voor respectievelijk tweemaal daags 5 mg en 10 mg). Voor patiënten op monotherapie (totaal 1.750) bedroegen de aantallen 48,9 en 41,9 patiënten met voorvallen per 100 patiëntjaren voor respectievelijk tweemaal daags 5 mg en 10 mg. Voor patiënten op achtergrond-DMARD's (totaal 3.117) bedroegen de aantallen 41,0 en 50,3 patiënten met voorvallen per 100 patiëntjaren voor respectievelijk tweemaal daags 5 mg en 10 mg.
Spondylitis ankylopoetica
In de gecombineerde klinische fase 2- en fase 3-onderzoeken, tijdens de placebogecontroleerde periode van maximaal 16 weken, bedroeg de frequentie van infecties in de groep met tweemaal daags 5 mg tofacitinib (185 patiënten) 27,6% en de frequentie in de placebogroep (187 patiënten) bedroeg 23,0%. In de gecombineerde klinische fase 2- en fase 3-onderzoeken bedroeg de frequentie van infecties 35,1% onder de 316 patiënten die gedurende maximaal 48 weken werden behandeld met tweemaal daags 5 mg tofacitinib.
Colitis ulcerosa
In de gerandomiseerde, 8 weken durende fase 2/3-inductieonderzoeken waren de percentages patiënten met infecties 21,1% (198 patiënten) in de groep met tweemaal daags 10 mg tofacitinib vergeleken met 15,2% (43 patiënten) in de placebogroep. In het gerandomiseerde, 52 weken durende fase 3-onderhoudsonderzoek waren de percentages patiënten met infecties 35,9% (71 patiënten) in de groep met tweemaal daags 5 mg tofacitinib en 39,8% (78 patiënten) in de groep met tweemaal daags 10 mg tofacitinib, vergeleken met 24,2% (48 patiënten) in de placebogroep.
In de gehele behandelervaring met tofacitinib was de vaakst gemelde infectie nasofaryngitis, hetgeen optrad bij 18,2% van de patiënten (211 patiënten).
In de gehele behandelervaring met tofacitinib was het totale incidentiecijfer voor infecties 60,3 voorvallen per 100 patiëntjaren (49,4% van de patiënten; totaal 572 patiënten).
Ernstige infecties
Reumatoïde artritis
In de 6 maanden en 24 maanden durende gecontroleerde klinische onderzoeken bedroeg het aantal ernstige infecties in de groep met tweemaal daags 5 mg tofacitinib als monotherapie 1,7 patiënt met voorvallen per 100 patiëntjaren. In de groep met tweemaal daags 10 mg tofacitinib als monotherapie bedroeg het aantal 1,6 patiënt met voorvallen per 100 patiëntjaren, het aantal bedroeg 0 voorvallen per 100 patiëntjaren voor de placebogroep en het aantal bedroeg 1,9 patiënt met voorvallen per 100 patiëntjaren voor de MTX-groep.
In onderzoeken met een duur van 6, 12 of 24 maanden bedroegen de aantallen ernstige infecties in de groep met tweemaal daags 5 mg tofacitinib plus DMARD en in de groep met tweemaal daags 10 mg tofacitinib plus DMARD respectievelijk 3,6 en 3,4 patiënten met voorvallen per 100 patiëntjaren, vergeleken met 1,7 patiënt met voorvallen per 100 patiëntjaren in de groep met placebo plus DMARD.
In de langetermijnveiligheidspopulatie met alle blootstellingen bedroegen de totale aantallen ernstige infecties 2,4 en 3,0 patiënten met voorvallen per 100 patiëntjaren voor respectievelijk de groep met tweemaal daags 5 mg en de groep met tweemaal daags 10 mg tofacitinib. De vaakst voorkomende ernstige infecties waren pneumonie, herpes zoster, urineweginfectie, cellulitis, gastro-enteritis en diverticulitis. Gevallen van opportunistische infecties zijn gemeld (zie rubriek 4.4).
In een groot (N=4.362), gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, werd een dosisafhankelijke toename van ernstige infecties waargenomen met tofacitinib vergeleken met TNF‑remmers (zie rubriek 4.4).
De incidentiecijfers (95%‑BI) voor ernstige infecties voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers bedroegen respectievelijk 2,86 (2,41; 3,37), 3,64 (3,11; 4,23), en 2,44 (2,02; 2,92) patiënten met voorvallen per 100 patiëntjaren. Vergeleken met TNF‑remmers was de hazardratio (HR) voor ernstige infecties 1,17 (0,92; 1,50) en 1,48 (1,17; 1,87) voor respectievelijk tweemaal daags 10 mg tofacitinib en tweemaal daags 5 mg tofacitinib.
Spondylitis ankylopoetica
In de gecombineerde klinische fase 2- en fase 3-onderzoeken kwam onder de 316 patiënten die gedurende maximaal 48 weken werden behandeld met tweemaal daags 5 mg tofacitinib, één ernstige infectie (aseptische meningitis) voor, wat neerkomt op 0,43 patiënten met voorvallen per 100 patiëntjaren.
Colitis ulcerosa
De incidentiecijfers en typen ernstige infecties in de klinische onderzoeken naar UC waren over het algemeen vergelijkbaar met de incidentiecijfers en typen ernstige infecties die werden gemeld in klinische onderzoeken naar RA met behandelgroepen met tofacitinib als monotherapie.
Ernstige infecties bij ouderen
Van de 4.271 patiënten die deelnamen aan RA-onderzoeken I–VI (zie rubriek 5.1) waren in totaal 608 RA-patiënten 65 jaar en ouder, waaronder 85 patiënten van 75 jaar en ouder. De frequentie van ernstige infecties bij met tofacitinib behandelde patiënten van 65 jaar en ouder was hoger dan bij degenen jonger dan 65 jaar (respectievelijk 4,8 per 100 patiëntjaren versus 2,4 per 100 patiëntjaren).
In een groot (N=4.362), gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, werd een toename van ernstige infecties waargenomen bij patiënten van 65 jaar en ouder voor tweemaal daags 10 mg tofacitinib vergeleken met TNF‑remmers en met tweemaal daags 5 mg tofacitinib (zie rubriek 4.4). De incidentiecijfers (95%‑BI) voor ernstige infecties bij patiënten van 65 jaar en ouder waren 4,03 (3,02; 5,27), 5,85 (4,64; 7,30) en 3,73 (2,81; 4,85) patiënten met voorvallen per 100 patiëntjaren voor respectievelijk tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib, en TNF‑remmers.
Vergeleken met TNF-remmers was de hazardratio (HR) voor ernstige infecties bij patiënten van 65 jaar en ouder 1,08 (0,74; 1,58) en 1,55 (1,10; 2,19) voor respectievelijk tweemaal daags 5 mg tofacitinib en tweemaal daags 10 mg tofacitinib.
Ernstige infecties uit niet-interventioneel veiligheidsonderzoek na goedkeuring
Uit gegevens uit een niet-interventioneel veiligheidsonderzoek na goedkeuring, waarin tofacitinib werd geëvalueerd bij RA‑patiënten uit een register (US Corrona), is gebleken dat voor de 11 mg tablet met verlengde afgifte, eenmaal daags toegediend, een numeriek hoger incidentiepercentage van ernstige infectie werd waargenomen dan voor de 5 mg filmomhulde tablet, tweemaal daags toegediend. Onbewerkte incidentiepercentages (95%‑BI) (d.w.z. niet gecorrigeerd voor leeftijd of geslacht) op basis van de beschikbaarheid van elke formulering waren op 12 maanden na aanvang van de behandeling 3,45 (1,93; 5,69) en 2,78 (1,74; 4,21) en op 36 maanden 4,71 (3,08; 6,91) en 2,79 (2,01; 3,77) patiënten met voorvallen per 100 patiëntjaren in respectievelijk de groep met eenmaal daags een 11 mg tablet met verlengde afgifte en de groep met tweemaal daags een 5 mg filmomhulde tablet. De niet-gecorrigeerde hazardratio was 1,30 (95%‑BI: 0,67; 2,50) op 12 maanden en 1,93 (95%‑BI: 1,15; 3,24) op 36 maanden, voor de eenmaaldaagse dosis van de 11 mg tablet met verlengde afgifte, vergeleken met de tweemaaldaagse dosis van de 5 mg filmomhulde tablet. De gegevens zijn gebaseerd op een klein aantal patiënten, met voorvallen die werden waargenomen met relatief grote betrouwbaarheidsintervallen en beperkte follow-upperiode.
Virale reactivering
Patiënten die worden behandeld met tofacitinib die van Japanse of Koreaanse afkomst zijn, of patiënten met langbestaande RA die eerder al behandeld zijn met twee of meer biologische DMARD's, of patiënten met een ALC van minder dan 1.000 cellen/mm3, of patiënten die worden behandeld met tweemaal daags 10 mg, kunnen een verhoogd risico hebben op herpes zoster (zie rubriek 4.4).
In een grootschalig (N=4.362) gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, werd een toename van voorvallen van herpes zoster waargenomen bij patiënten die werden behandeld met tofacitinib vergeleken met TNF-remmers. De incidentiepercentages (95%-BI) voor herpes zoster voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers bedroegen respectievelijk 3,75 (3,22; 4,34), 3,94 (3,38; 4,57) en 1,18 (0,90; 1,52) patiënten met voorvallen per 100 patiëntjaren.
Laboratoriumtesten
Lymfocyten
In de gecontroleerde klinische onderzoeken naar RA werden ALC-dalingen tot minder dan 500 cellen/mm3 bevestigd bij 0,3% van de patiënten en voor ALC tussen 500 en 750 cellen/mm3 bij 1,9% van de patiënten voor de doses van tweemaal daags 5 mg en tweemaal daags 10 mg gecombineerd.
In de langetermijnveiligheidspopulatie met RA werden ALC-dalingen tot minder dan 500 cellen/mm3 bevestigd bij 1,3% van de patiënten en voor ALC tussen 500 en 750 cellen/mm3 bij 8,4% van de patiënten voor de doses van tweemaal daags 5 mg en tweemaal daags 10 mg gecombineerd.
Een bevestigde ALC van minder dan 750 cellen/mm3 ging gepaard met een verhoogde incidentie van ernstige infecties (zie rubriek 4.4).
In de klinische onderzoeken naar UC waren de veranderingen in ALC die werden waargenomen bij behandeling met tofacitinib vergelijkbaar met de veranderingen die werden waargenomen in klinische onderzoeken naar RA.
Neutrofielen
In de gecontroleerde klinische onderzoeken naar RA werden dalingen in de ANC tot minder dan 1.000 cellen/mm3 bevestigd bij 0,08% van de patiënten voor de doses van tweemaal daags 5 mg en tweemaal daags 10 mg gecombineerd. In geen enkele behandelgroep werden bevestigde dalingen in de ANC tot minder dan 500 cellen/mm3 waargenomen. Er was geen duidelijk verband tussen neutropenie en het optreden van ernstige infecties.
In de langetermijnveiligheidspopulatie met RA bleven het patroon en de incidentie van bevestigde dalingen in de ANC overeenkomen met wat werd gezien in de gecontroleerde klinische onderzoeken (zie rubriek 4.4).
In de klinische onderzoeken naar UC waren de veranderingen in ANC die werden waargenomen bij behandeling met tofacitinib vergelijkbaar met de veranderingen die werden waargenomen in klinische onderzoeken naar RA.
Trombocyten
Patiënten in de gecontroleerde klinische fase 3-onderzoeken (RA, PsA, SA, CU) dienden een trombocytentelling van ≥ 100.000 cellen/mm3 te hebben om in aanmerking te komen voor deelname aan het onderzoek. Daarom is er geen informatie beschikbaar over patiënten die vóór aanvang van de behandeling met tofacitinib een trombocytentelling < 100.000 cellen/mm3 hadden.
Leverenzymtesten
Bevestigde verhogingen in leverenzymen groter dan 3 keer de bovengrens van de normaalwaarde (3x ULN) werden soms waargenomen bij RA-patiënten. Bij patiënten met verhoogde leverenzymwaarden resulteerde een wijziging in het behandelregime, zoals een verlaging van de dosis van gelijktijdige DMARD, onderbreking van tofacitinib of een verlaging van de dosis tofacitinib, tot een daling of normalisatie van de leverenzymwaarden.
In het gecontroleerde gedeelte van het fase 3-monotherapieonderzoek (0–3 maanden) naar RA (onderzoek I, zie rubriek 5.1) werden ALAT-stijgingen groter dan 3x ULN waargenomen bij 1,65%, 0,41% en 0% van de patiënten die respectievelijk placebo, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen. In dit onderzoek werden ASAT-stijgingen groter dan 3x ULN waargenomen bij 1,65%, 0,41% en 0% van de patiënten die respectievelijk placebo, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen.
In het fase 3-monotherapieonderzoek (0–24 maanden) naar RA (onderzoek VI, zie rubriek 5.1) werden ALAT-stijgingen groter dan 3x ULN waargenomen bij 7,1%, 3,0% en 3,0% van de patiënten die respectievelijk methotrexaat, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen. In dit onderzoek werden ASAT-stijgingen groter dan 3x ULN waargenomen bij 3,3%, 1,6% en 1,5% van de patiënten die respectievelijk methotrexaat, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen.
In het gecontroleerde gedeelte van de fase 3-onderzoeken naar RA met achtergrond-DMARD's (0–3 maanden) (onderzoek II–V, zie rubriek 5.1) werden ALAT-stijgingen groter dan 3x ULN waargenomen bij 0,9%, 1,24% en 1,14% van de patiënten die respectievelijk placebo, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen. In deze onderzoeken werden ASAT-stijgingen groter dan 3x ULN waargenomen bij 0,72%, 0,5% en 0,31% van de patiënten die respectievelijk placebo, tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen.
In de langetermijnextensieonderzoeken naar RA met monotherapie werden ALAT-stijgingen groter dan 3x ULN waargenomen bij 1,1% en 1,4% van de patiënten die respectievelijk tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen. ASAT-stijgingen groter dan 3x ULN werden waargenomen bij < 1,0% in zowel de groep met tweemaal daags 5 mg tofacitinib als in de groep met tweemaal daags 10 mg tofacitinib.
In de langetermijnextensieonderzoeken naar RA met achtergrond-DMARD's werden ALAT-stijgingen groter dan 3x ULN waargenomen bij 1,8% en 1,6% van de patiënten die respectievelijk tweemaal daags 5 mg en tweemaal daags 10 mg tofacitinib kregen. ASAT-stijgingen groter dan 3x ULN werden waargenomen bij < 1,0% in zowel de groep met tweemaal daags 5 mg tofacitinib als in de groep met tweemaal daags 10 mg tofacitinib.
In een grootschalig (N=4.362) gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, werden ALAT-stijgingen groter dan of gelijk aan 3x ULN waargenomen bij 6,01%, 6,54% en 3,77% van de patiënten die respectievelijk tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers kregen. ASAT-stijgingen groter dan of gelijk aan 3x ULN werden waargenomen bij 3,21%, 4,57% en 2,38% van de patiënten die respectievelijk tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers kregen.
In de klinische onderzoeken naar UC waren de veranderingen in leverenzymtesten die werden waargenomen bij behandeling met tofacitinib vergelijkbaar met de veranderingen die werden waargenomen in klinische onderzoeken naar RA.
Lipiden
Stijgingen in lipidenparameters (totaal cholesterol, LDL-cholesterol, HDL-cholesterol, triglyceriden) werden voor de eerste keer beoordeeld 1 maand na aanvang van tofacitinib in de gecontroleerde, dubbelblinde klinische onderzoeken bij RA. Op dit tijdpunt werden stijgingen waargenomen die daarna stabiel bleven.
De veranderingen in lipidenparameters vanaf de beginmeting tot en met het einde van het onderzoek (6–24 maanden) in de gecontroleerde klinische onderzoeken bij RA worden hieronder samengevat:
- De gemiddelde LDL-cholesterolwaarde steeg met 15% in de arm met tweemaal daags 5 mg tofacitinib en met 20% in de arm met tweemaal daags 10 mg tofacitinib na 12 maanden, en steeg met 16% in de arm met tweemaal daags 5 mg tofacitinib en met 19% in de arm met tweemaal daags 10 mg tofacitinib na 24 maanden.
- De gemiddelde HDL-cholesterolwaarde steeg met 17% in de arm met tweemaal daags 5 mg tofacitinib en met 18% in de arm met tweemaal daags 10 mg tofacitinib na 12 maanden, en steeg met 19% in de arm met tweemaal daags 5 mg tofacitinib en met 20% in de arm met tweemaal daags 10 mg tofacitinib na 24 maanden.
Na het staken van de behandeling met tofacitinib keerden de lipidenwaarden terug naar de beginwaarden.
De gemiddelde verhoudingen LDL-cholesterol/HDL-cholesterol en apolipoproteïne B (ApoB)/ApoA1 waren in principe onveranderd bij met tofacitinib behandelde patiënten.
In een gecontroleerd klinisch onderzoek naar RA namen de verhoogde LDL-cholesterol- en ApoB-waarden weer af naar de waarden van voor de behandeling als reactie op behandeling met statines.
In de langetermijnveiligheidspopulaties met RA bleven de stijgingen in lipidenparameters overeenkomen met wat werd gezien in de gecontroleerde klinische onderzoeken.
De veranderingen in lipidenparameters, gemeten vanaf de bepaling van de uitgangswaarden tot en met 24 maanden daarna in een grootschalig (N=4.362) gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, worden hieronder samengevat:
- De stijging van de gemiddelde LDL-cholesterolwaarde bedroeg na 12 maanden 13,80%, 17,04% en 5,50% bij patiënten die respectievelijk tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en een TNF-remmer kregen. Na 24 maanden bedroeg de stijging respectievelijk 12,71%, 18,14% en 3,64%.
- De stijging van de gemiddelde HDL-cholesterolwaarde bedroeg na 12 maanden 11,71%, 13,63% en 2,82% bij patiënten die respectievelijk tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en een TNF-remmer kregen. Na 24 maanden bedroeg de stijging respectievelijk 11,58%; 13,54% en 1,42%.
In de klinische onderzoeken naar UC waren de veranderingen in lipiden die werden waargenomen bij behandeling met tofacitinib vergelijkbaar met de veranderingen die werden waargenomen in klinische onderzoeken naar RA.
Myocardinfarct
Reumatoïde artritis
In een groot (N=4.362) gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, bedroegen de incidentiecijfers (95%-BI) voor niet-fataal myocardinfarct voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers respectievelijk 0,37 (0,22; 0,57), 0,33 (0,19; 0,53) en 0,16 (0,07; 0,31) per 100 patiëntjaren. Er werden enkele fatale myocardinfarcten gemeld met vergelijkbare cijfers bij patiënten behandeld met tofacitinib ten opzichte van patiënten behandeld met TNF-remmers (zie rubriek 4.4 en 5.1). Het onderzoek vereiste dat ten minste 1.500 patiënten gedurende 3 jaar werden gevolgd.
Maligniteiten met uitzondering van NMSC
Reumatoïde artritis
In een groot (N=4.362) gerandomiseerd postmarketingveiligheidsonderzoek bij patiënten met RA van 50 jaar of ouder met ten minste één bijkomende cardiovasculaire risicofactor, bedroegen de incidentiecijfers (95%-BI) voor longkanker voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers respectievelijk 0,23 (0,12; 0,40), 0,32 (0,18; 0,51) en 0,13 (0,05; 0,26) per 100 patiëntjaren (zie rubriek 4.4 en 5.1). Het onderzoek vereiste dat ten minste 1.500 patiënten gedurende 3 jaar werden gevolgd.
De incidentiecijfers (95%-BI) voor lymfoom bedroegen voor tweemaal daags 5 mg tofacitinib, tweemaal daags 10 mg tofacitinib en TNF-remmers respectievelijk 0,07 (0,02; 0,18), 0,11 (0,04; 0,24) en 0,02 (0,00; 0,10) per 100 patiëntjaren (zie rubriek 4.4 en 5.1).
Pediatrische patiënten
Polyarticulaire juveniele idiopathische artritis en juveniele PsA
De bijwerkingen bij JIA‑patiënten in het klinische ontwikkelingsprogramma waren wat betreft type en frequentie in overeenstemming met de bijwerkingen die werden gezien bij volwassen RA‑patiënten, met uitzondering van enkele infecties (griep, faryngitis, sinusitis, virale infectie) en gastro-intestinale of algemene aandoeningen (buikpijn, nausea, braken, pyrexie, hoofdpijn, hoesten). Deze kwamen vaker voor bij pediatrische patiënten met JIA. MTX was de csDMARD die het vaakst gelijktijdig werd gebruikt (op dag 1 namen 156 van 157 patiënten die csDMARD’s gebruikten, MTX). Er zijn onvoldoende gegevens beschikbaar over het veiligheidsprofiel van tofacitinib wanneer dit gelijktijdig met andere csDMARD’s wordt gebruikt.
Infecties
In het dubbelblinde deel van het fase 3-hoofdonderzoek (onderzoek JIA‑I) was infectie de vaakst gemelde bijwerking (44,3%). De infecties waren over het algemeen licht tot matig van aard.
In de geïntegreerde veiligheidspopulatie hadden 7 patiënten ernstige infecties tijdens behandeling met tofacitinib binnen de meldingsperiode (tot maximaal 28 dagen na de laatste dosis onderzoeksgeneesmiddel). Dit gaf een incidentiecijfer van 1,92 patiënten met voorvallen per 100 patiëntjaren: pneumonie, epiduraal empyeem (met sinusitis en subperiostaal abces), pilonidaliscyste, appendicitis, Escherichia-pyelonefritis, abces ledemaat en urineweginfectie.
In de geïntegreerde veiligheidspopulatie hadden 3 patiënten niet-ernstige voorvallen van herpes zoster binnen het meldingsvenster. Dit gaf een incidentiecijfer van 0,82 patiënten met voorvallen per 100 patiëntjaren weer. Nog één (1) andere patiënt had een voorval van ernstige herpes zoster buiten het meldingsvenster.
Hepatische voorvallen
Patiënten in het JIA‑hoofdonderzoek moesten ASAT- en ALAT-waarden lager dan 1,5 maal de bovengrens van normaal (ULN, Upper Limit of Normal) hebben om in aanmerking te komen voor deelname aan het onderzoek. In de geïntegreerde veiligheidspopulatie waren er 2 patiënten die bij 2 opeenvolgende bezoeken verhoogde ALAT-waarden van ≥ 3 maal de ULN hadden. Geen van de voorvallen voldeed aan de criteria van de wet van Hy. Beide patiënten kregen MTX als achtergrondbehandeling en elk voorval herstelde na stopzetting van MTX en blijvende stopzetting van tofacitinib.
Laboratoriumtesten
Veranderingen in resultaten van laboratoriumtesten bij JIA‑patiënten in het klinische ontwikkelingsprogramma kwamen overeen met de veranderingen die werden gezien bij volwassen RA‑patiënten. Patiënten in het JIA‑hoofdonderzoek moesten een trombocytentelling van ≥ 100.000 cellen/mm3 hebben om in aanmerking te komen voor deelname aan het onderzoek. Daarom is er geen informatie beschikbaar over JIA‑patiënten die vóór aanvang van de behandeling met tofacitinib een trombocytentelling < 100.000 cellen/mm3 hadden.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg worden verzocht alle vermoedelijke bijwerkingen te melden via:
België: Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
E-mail: adr@fagg-afmps.be
Nederland: Nederlands Bijwerkingen Centrum Lareb
Website: www.lareb.nl.
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Brussel
België
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/17/1178/001
EU/1/17/1178/002
EU/1/17/1178/003
EU/1/17/1178/004
EU/1/17/1178/005
EU/1/17/1178/006
EU/1/17/1178/007
EU/1/17/1178/008
EU/1/17/1178/009
EU/1/17/1178/014
10. DATUM VAN HERZIENING VAN DE TEKST
09/01/26
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
26A09
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3558210 | XELJANZ 5MG FILMOMH TABL 56 X 5MG BLISTER | L04AA29 | € 883,74 | - | Ja | € 12,8 | € 8,5 |
| 3558244 | XELJANZ 5MG FILMOMH TABL 180 X 5MG FLACON | L04AA29 | € 2444,22 | - | Ja | € 15,9 | € 10,5 |
| 3611613 | XELJANZ 5MG FILMOMH TABL 182 X 5MG BLISTER | L04AA29 | € 2471,26 | - | Ja | € 15,9 | € 10,5 |
| 3786415 | XELJANZ 10MG FILMOMH TABL 112 X 10MG BLISTER | L04AA29 | € 2711,88 | - | Ja | € 15,9 | € 10,5 |
| 3786423 | XELJANZ 10MG FILMOMH TABL 56 X 10MG BLISTER | L04AA29 | € 1361,42 | - | Ja | € 12,8 | € 8,5 |
| 3809803 | XELJANZ 5MG FILMOMH TABL 112 X 5MG BLISTER | L04AA29 | € 1361,42 | - | Ja | € 15,9 | € 10,5 |