SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Forxiga 5 mg, filmomhulde tabletten
Forxiga 10 mg, filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Forxiga 5 mg, filmomhulde tabletten
Elke tablet bevat dapagliflozinepropaandiolmonohydraat, overeenkomend met 5 mg dapagliflozine.
Hulpstof met bekend effect
Elke 5 mg tablet bevat 25 mg lactose.
Forxiga 10 mg, filmomhulde tabletten
Elke tablet bevat dapagliflozinepropaandiolmonohydraat, overeenkomend met 10 mg dapagliflozine.
Hulpstof met bekend effect
Elke 10 mg tablet bevat 50 mg lactose.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet)
Forxiga 5 mg, filmomhulde tabletten
Gele, biconvexe, ronde (diameter 0,7 cm), filmomhulde tabletten met aan de ene zijde “5” in reliëf en aan de andere zijde “1427” in reliëf.
Forxiga 10 mg, filmomhulde tabletten
Gele, biconvexe, ongeveer 1,1 x 0,8 cm diagonaal diamantvormige, filmomhulde tabletten met aan de ene zijde “10” in reliëf en aan de andere zijde “1428” in reliëf.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Diabetes mellitus type 2
Forxiga is geïndiceerd voor gebruik bij volwassenen en kinderen van 10 jaar en ouder voor de behandeling van onvoldoende gereguleerde diabetes mellitus type 2 als aanvulling op dieet en lichaamsbeweging
- als monotherapie wanneer metformine ongeschikt wordt geacht vanwege onverdraagbaarheid.
- als aanvulling op andere geneesmiddelen voor de behandeling van diabetes type 2.
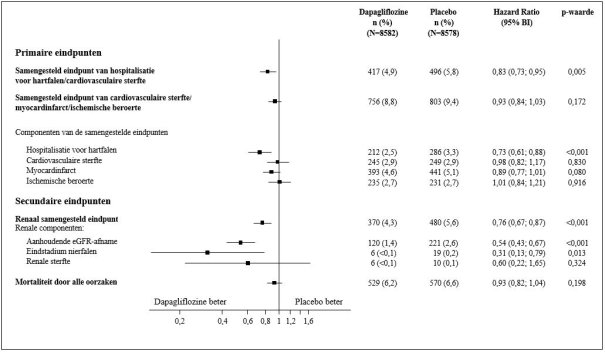
Voor onderzoeksresultaten betreffende combinatie van therapieën, effecten op de bloedglucoseregulatie, cardiovasculaire en renale voorvallen en de onderzochte populaties, zie de rubrieken 4.4, 4.5 en 5.1.
Hartfalen
Forxiga is geïndiceerd voor gebruik bij volwassenen voor de behandeling van symptomatisch chronisch hartfalen.
Chronische nierschade
Forxiga is geïndiceerd voor gebruik bij volwassenen voor de behandeling van chronische nierschade.
4.2 Dosering en wijze van toediening
Dosering
Diabetes mellitus type 2
De aanbevolen dosering is 10 mg dapagliflozine eenmaal daags.
Wanneer dapagliflozine wordt gebruikt in combinatie met insuline of een insulinesecretie-bevorderend middel, zoals een sulfonylureumderivaat, kan een lagere dosering insuline of insulinesecretie-bevorderend middel worden overwogen om het risico op hypoglykemie te verminderen (zie rubrieken 4.5 en 4.8).
Hartfalen
De aanbevolen dosering is 10 mg dapagliflozine eenmaal daags.
Chronische nierschade
De aanbevolen dosering is 10 mg dapagliflozine eenmaal daags.
Speciale patiëntengroepen
Nierinsufficiëntie
Er is geen nierfunctie-afhankelijke dosisaanpassing vereist.
Vanwege de beperkte ervaring wordt het niet aanbevolen om een behandeling met dapagliflozine te starten bij patiënten met een GFR < 25 ml/min.
Bij patiënten met diabetes mellitus type 2 is de glucoseverlagende werkzaamheid van dapagliflozine verminderd wanneer de glomerulaire filtratiesnelheid (GFR) < 45 ml/min bedraagt en is waarschijnlijk afwezig bij patiënten met ernstige nierinsufficiëntie. Als de GFR onder 45 ml/min daalt, moet daarom bij patiënten met diabetes mellitus type 2 een aanvullende glucoseverlagende behandeling worden overwogen als verdere glykemische controle nodig is (zie rubrieken 4.4, 4.8, 5.1 en 5.2).
Leverinsufficiëntie
Er is geen dosisaanpassing nodig bij patiënten met een lichte of matige leverfunctiestoornis. Bij patiënten met een ernstige leverfunctiestoornis wordt een startdosis van 5 mg aangeraden. Indien deze goed wordt verdragen kan de dosis worden verhoogd naar 10 mg (zie rubrieken 4.4 en 5.2).
Ouderen (≥ 65 jaar)
Er wordt geen leeftijdsafhankelijke dosisaanpassing aanbevolen.
Pediatrische patiënten
Er is geen dosisaanpassing nodig voor de behandeling van diabetes mellitus type 2 bij kinderen van 10 jaar en ouder (zie rubriek 5.1 en 5.2). Er zijn geen gegevens beschikbaar over kinderen jonger dan 10 jaar.
De veiligheid en werkzaamheid van dapagliflozine voor de behandeling van hartfalen of de behandeling van chronische nierschade bij kinderen in de leeftijd tot 18 jaar zijn nog niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Forxiga kan eenmaal daags oraal worden ingenomen op ieder moment van de dag, met of zonder voedsel. De tabletten moeten in het geheel worden doorgeslikt.
4.3 Contra-indicaties
Overgevoeligheid voor het werkzame bestanddeel of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Diabetes mellitus type 2
In de klinische studies bij diabetes mellitus type 2 zijn meer dan 15.000 patiënten behandeld met dapagliflozine.
De primaire beoordeling van veiligheid en verdraagbaarheid werd uitgevoerd in een vooraf gespecificeerde, gepoolde analyse van 13 kortdurende (tot 24 weken) placebogecontroleerde studies met 2.360 proefpersonen behandeld met dapagliflozine 10 mg en 2.295 proefpersonen behandeld met placebo.
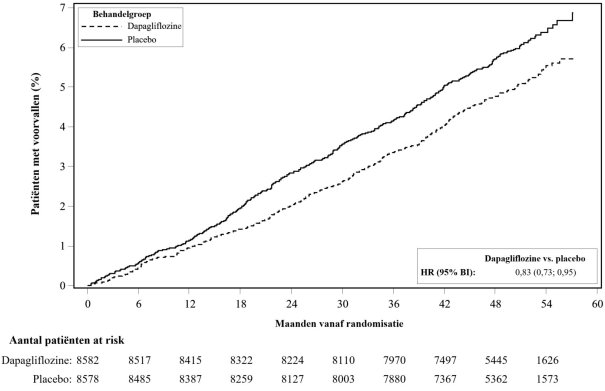
In de studie naar cardiovasculaire uitkomsten voor dapagliflozine bij diabetes mellitus type 2 (DECLARE-studie, zie rubriek 5.1) ontvingen 8.574 patiënten dapagliflozine 10 mg en ontvingen 8.569 patiënten placebo gedurende een mediane blootstellingstijd van 48 maanden. In totaal waren er 30.623 patiëntjaren van blootstelling aan dapagliflozine.
De meest voorkomende gerapporteerde bijwerkingen in alle klinische studies waren genitale infecties.
Hartfalen
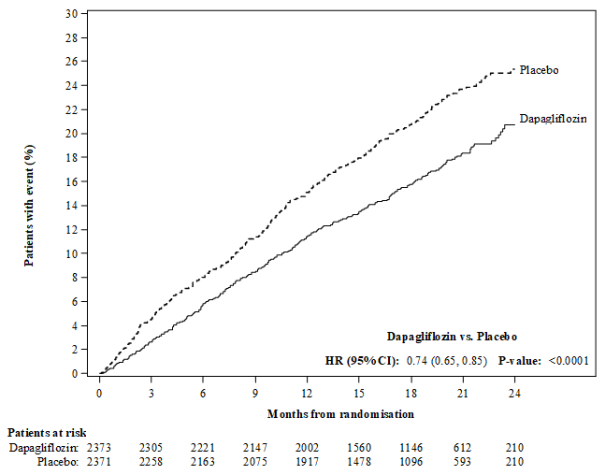
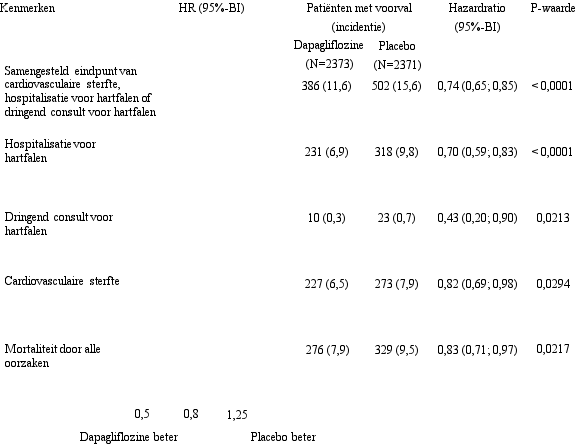
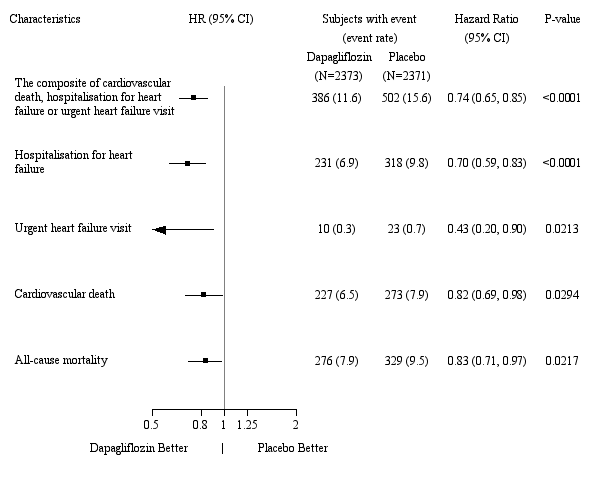
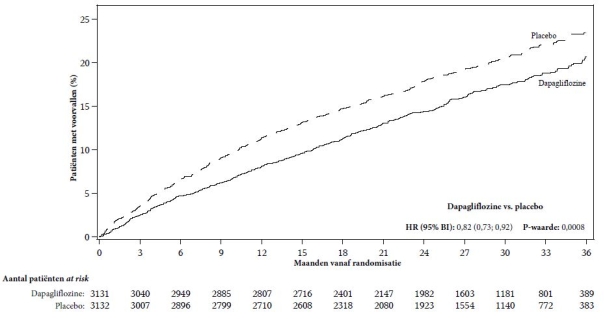
In de studie naar cardiovasculaire uitkomsten voor dapagliflozine bij patiënten met hartfalen met verminderde ejectiefractie (DAPA-HF-studie) werden 2.368 patiënten behandeld met dapagliflozine 10 mg en 2.368 patiënten met placebo met een mediane blootstellingsduur van 18 maanden. De patiëntenpopulatie bestond uit patiënten met diabetes mellitus type 2 en patiënten zonder diabetes en patiënten met eGFR ≥ 30 ml/min/1,73 m2. In de studie naar cardiovasculaire uitkomsten voor dapagliflozine bij patiënten met hartfalen met linkerventrikelejectiefractie > 40% (DELIVER), werden 3.126 patiënten behandeld met dapagliflozine 10 mg en 3.127 patiënten met placebo met een mediane blootstellingsduur van 27 maanden. De patiëntenpopulatie bestond uit patiënten met diabetes mellitus type 2 en zonder diabetes, en patiënten met eGFR ≥ 25 ml/min/1,73 m2.
Het algemene veiligheidsprofiel van dapagliflozine bij patiënten met hartfalen was consistent met het bekende veiligheidsprofiel van dapagliflozine.
Chronische nierschade
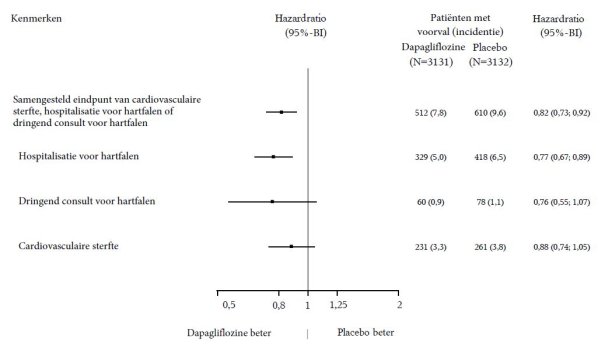
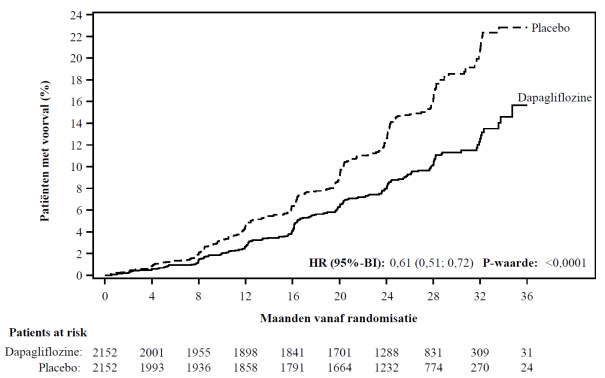
In de studie naar renale uitkomsten voor dapagliflozine bij patiënten met chronische nierschade (DAPA-CKD) werden 2.149 patiënten behandeld met dapagliflozine 10 mg en 2.149 patiënten met placebo met een mediane blootstellingsduur van 27 maanden. De patiëntenpopulatie bestond uit patiënten met diabetes mellitus type 2 en patiënten zonder diabetes, met een eGFR ≥ 25 tot ≤ 75 ml/min/1,73 m2 en albuminurie (urinealbumine creatinineverhouding [UACR] ≥ 200 en ≤ 5.000 mg/g). De behandeling werd voortgezet als de eGFR daalde tot een waarde lager dan 25 ml/min/1,73 m2.
Het algemene veiligheidsprofiel van dapagliflozine bij patiënten met chronische nierschade was consistent met het bekende veiligheidsprofiel van dapagliflozine.
Bijwerkingen in tabelvorm
De volgende bijwerkingen zijn vastgesteld op basis van de placebogecontroleerde klinische studies en post-marketingsurveillance. Geen enkele bijwerking werd dosisafhankelijk bevonden. De onderstaande bijwerkingen zijn geclassificeerd naar frequentie en Systeem/Orgaanklasse (SOK). De frequentiecategorieën zijn als volgt gedefinieerd: zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald).
Tabel 1: Bijwerkingen uit placebogecontroleerde klinische studiesa en post-marketingervaring
Systeem/ Orgaanklasse | Zeer vaak | Vaak* | Soms ** | Zelden | Zeer zelden |
Infecties en parasitaire aandoeningen |
| Vulvovaginitis, balanitis en gerelateerde genitale infecties*,b,c | Schimmel-infectie** |
| Necrotise-rende fasciitis van het perineum (fournier-gangreen)b, i |
Voedings- en stofwisselings-stoornissen | Hypoglykemie (bij gebruik met SU of insuline)b |
| Volume-depletieb,e | Diabetische ketoacidose (wanneer gebruikt bij diabetes mellitus type 2)b,i,k |
|
Zenuwstelsel-aandoeningen |
| Duizeligheid |
|
|
|
Maagdarmstelsel-aandoeningen |
|
| Obstipatie** |
|
|
Huid- en onderhuid-aandoeningen |
| Rashj |
|
| Angio-oedeem |
Skeletspierstelsel- en bindweefsel-aandoeningen |
| Rugpijn* |
|
|
|
Nier- en urineweg-aandoeningen |
| Dysurie | Nycturie** |
| Tubulo-interstitiële nefritis |
Voortplantings-stelsel- en borst-aandoeningen |
|
| Vulvovaginale pruritus** |
|
|
Onderzoeken |
| Verhoogd hematocrietg | Verhoogd bloedcreatinine gedurende initiële behandeling**,b |
|
|
a De tabel toont gegevens tot 24 weken (korte termijn) ongeacht het gebruik van glykemische noodmedicatie.
b Zie de bijbehorende subrubriek hieronder voor verdere informatie.
c Vulvovaginitis, balanitis en soortgelijke genitale infecties omvatten bijvoorbeeld de vooraf gedefinieerde voorkeurstermen: vulvovaginale schimmelinfectie, vaginale infectie, balanitis, genitale schimmelinfectie, vulvovaginale candidiasis, vulvovaginitis, balanitis candida, genitale candidiase, genitale infectie, genitale infectie bij mannen, penisinfectie, vulvitis, bacteriële vaginitis, abces in de vulva.
d Urineweginfectie omvat de volgende voorkeurstermen, op volgorde van gemelde frequentie: urineweginfectie, cystitis, urineweginfectie met Escherichia, genito-urinaire infectie, pyelonefritis, trigonitis, uretritis, nierinfectie en prostatitis.
e Volumedepletie omvat de vooraf gedefinieerde voorkeurstermen: dehydratatie, hypovolemie, hypotensie.
f Polyurie omvat de voorkeurstermen: pollakisurie, polyurie, verhoogde urineproductie.
g Gemiddelde veranderingen ten opzichte van de baseline in hematocriet waren 2,30% voor dapagliflozine 10 mg versus ‑0,33% voor placebo. Hematocrietwaarden > 55% werden gemeld bij 1,3% van de proefpersonen die met dapagliflozine 10 mg werden behandeld versus 0,4% bij proefpersonen die placebo kregen.
h Gemiddelde procentuele verandering ten opzichte van de baseline voor dapagliflozine 10 mg versus placebo was respectievelijk: totaal cholesterol 2,5% versus 0,0%; HDL-cholesterol 6,0% versus 2,7%; LDL‑cholesterol 2,9% versus ‑1,0%; triglyceriden ‑2,7% versus ‑0,7%.
i Zie rubriek 4.4.
j Bijwerking werd door post-marketingsurveillance geïdentificeerd. Rash omvat de volgende voorkeurstermen, weergegeven op volgorde van frequentie in klinische studies: rash, gegeneraliseerde rash, jeukende rash, vlekkerige rash, maculopapilaire rash, pustuleuze rash, vesiculaire rash en erythemateuze rash. In actief- en placebogecontroleerde klinische studies (dapagliflozine, N=5936, alle controles, N=3403) was de frequentie van rash vergelijkbaar voor respectievelijk dapagliflozine (1,4%) en alle controles (1,4%).
k Gerapporteerd in de studie naar cardiovasculaire uitkomsten bij patiënten met diabetes type 2 (DECLARE). Frequentie is gebaseerd op jaarlijks percentage.
* Gerapporteerd bij ≥ 2% proefpersonen die met dapagliflozine 10 mg werden behandeld en ≥ 1% meer proefpersonen die met dapagliflozine 10 mg werden behandeld ten opzichte van placebo en ten minste 3 proefpersonen meer die met dapagliflozine 10 mg werden behandeld ten opzichte van placebo.
** Gerapporteerd door de onderzoeker als mogelijk gerelateerd, waarschijnlijk gerelateerd of gerelateerd aan de studiebehandeling en gemeld bij ≥ 0,2% van de proefpersonen en ≥ 0,1% vaker en bij ten minste 3 proefpersonen meer die werden behandeld met dapagliflozine 10 mg ten opzichte van placebo.
Beschrijving van geselecteerde bijwerkingen
Vulvovaginitis, balanitis en verwante genitale infecties
In de gepoolde veiligheidsdata van 13 studies werden vulvovaginitis, balanitis en soortgelijke genitale infecties gemeld bij 5,5% en 0,6% van de proefpersonen die respectievelijk dapagliflozine 10 mg en placebo kregen. De meeste infecties waren mild tot matig en proefpersonen reageerden op een initiële standaardbehandeling; de infecties leidden zelden tot staken van de dapagliflozinebehandeling. Deze infecties kwamen vaker voor bij vrouwen (8,4% voor dapagliflozine en 1,2% voor placebo) en proefpersonen met een voorgeschiedenis hadden een grotere kans om een terugkerende infectie te krijgen.
In de DECLARE-studie was het aantal patiënten met een genitale infectie als ernstige bijwerking laag en gebalanceerd: 2 patiënten in zowel de dapagliflozine- als de placebogroep.
In de DAPA-HF-studie meldde geen enkele patiënt ernstige bijwerkingen van genitale infecties in de dapagliflozinegroep, en één in de placebogroep. Er waren 7 (0,3%) patiënten met bijwerkingen die leidden tot stopzetting als gevolg van genitale infecties in de dapagliflozinegroep, en geen in de placebogroep. In de DELIVER-studie meldde één (< 0,1%) patiënt in elke behandelgroep een ernstige bijwerking van genitale infecties. Er waren 3 (0,1%) patiënten met bijwerkingen die leidden tot stopzetting als gevolg van genitale infecties in de dapagliflozinegroep en geen in de placebogroep.
In de DAPA-CKD-studie waren er 3 (0,1%) patiënten met ernstige bijwerkingen van genitale infecties in de dapagliflozinegroep en geen in de placebogroep. Er waren 3 (0,1%) patiënten met bijwerkingen die leidden tot stopzetting als gevolg van genitale infecties in de dapagliflozinegroep en geen in de placebogroep. Ernstige bijwerkingen van genitale infecties of bijwerkingen die tot stopzetting als gevolg van genitale infecties leidden, werden niet gemeld voor patiënten zonder diabetes.
Er zijn gevallen van fimose/verworven fimose gemeld met gelijktijdige genitale infecties; in sommige gevallen moest de patiënt worden besneden.
Necrotiserende fasciitis van het perineum (fournier-gangreen)
Na het in de handel brengen zijn er gevallen van fournier-gangreen gemeld bij patiënten die SGLT2-remmers innemen, waaronder dapagliflozine (zie rubriek 4.4).
In de DECLARE-studie met 17.160 patiënten met diabetes mellitus type 2 en een mediane blootstellingstijd van 48 maanden zijn in totaal 6 gevallen van fournier-gangreen gemeld; één in de groep behandeld met dapagliflozine en vijf in de placebogroep.
Hypoglykemie
De frequentie van hypoglykemie was afhankelijk van het soort achtergrondtherapie dat in de klinische studies met betrekking tot diabetes mellitus gebruikt werd.
In studies met dapagliflozine als monotherapie, als add-on combinatietherapie met metformine en als add-on combinatietherapie met sitagliptine (met of zonder metformine) was de frequentie van milde episodes van hypoglykemie vergelijkbaar (< 5%) in de behandelgroepen, inclusief placebo tot aan 102 weken van behandeling. In alle studies kwamen soms ernstige gevallen van hypoglykemie voor en de frequentie hiervan was vergelijkbaar voor de groepen die behandeld werden met dapagliflozine of met placebo. Studies met add-on sulfonylureumderivaten en een add-on insulinebehandeling vertoonden een hogere incidentie van hypoglykemie (zie rubriek 4.5).
In een add-on combinatiestudie met glimepiride werden op week 24 en 48 vaker milde episodes van hypoglykemie gerapporteerd in de groep die werd behandeld met dapagliflozine 10 mg plus glimepiride (resp. 6,0% en 7,9%) dan in de groep die werd behandeld met placebo plus glimepiride (resp. 2,1% en 2,1%).
In een add-on combinatiestudie met insuline werden episodes van ernstige hypoglykemie gerapporteerd bij 0,5% en 1,0% van de proefpersonen die werden behandeld met dapagliflozine 10 mg plus insuline in respectievelijk week 24 en week 104, en bij 0,5% van de proefpersonen die werden behandeld met placebo plus insuline in week 24 en week 104. In respectievelijk week 24 en week 104 werden milde episodes van hypoglykemie gerapporteerd bij 40,3% en 53,1% van de proefpersonen die dapagliflozine 10 mg plus insuline kregen en bij 34,0% en 41,6% van de proefpersonen die placebo plus insuline kregen.
In een add-on combinatiestudie met metformine en een sulfonylureumderivaat, gedurende 24 weken, werden geen episodes van ernstige hypoglykemie gerapporteerd. Minder ernstige episodes van hypoglykemie werden gerapporteerd bij 12,8% van de proefpersonen die dapagliflozine 10 mg plus metformine en een sulfonylureumderivaat kregen en bij 3,7% van de proefpersonen die placebo plus metformine en een sulfonylureumderivaat kregen.
In de DECLARE-studie werd geen verhoogd risico op ernstige hypoglykemie waargenomen bij dapagliflozinebehandeling vergeleken met placebo. Ernstige voorvallen van hypoglykemie werden gerapporteerd bij 58 (0,7%) van de patiënten behandeld met dapagliflozine en 83 (1,0%) van de patiënten behandeld met placebo.
In de DAPA-HF-studie werden ernstige voorvallen van hypoglykemie gemeld bij 4 (0,2%) patiënten in zowel de dapagliflozine- als placebogroep. In de DELIVER-studie werden ernstige voorvallen van hypoglykemie gemeld bij 6 (0,2%) patiënten in de dapagliflozinegroep en 7 (0,2%) patiënten in de placebogroep. Ernstige voorvallen van hypoglykemie werden enkel waargenomen bij patiënten met diabetes mellitus type 2.
In de DAPA-CKD-studie werden ernstige voorvallen van hypoglykemie gemeld bij 14 (0,7%) patiënten in de dapagliflozinegroep en bij 28 (1,3%) patiënten in de placebogroep en deze werden enkel waargenomen bij patiënten met diabetes mellitus type 2.
Volumedepletie
In de gepoolde veiligheidsdata van 13 studies werden bijwerkingen die zouden kunnen wijzen op volumedepletie (inclusief meldingen van dehydratatie, hypovolemie of hypotensie) gerapporteerd bij 1,1% en 0,7% van de proefpersonen die respectievelijk dapagliflozine 10 mg en placebo kregen. Ernstige bijwerkingen deden zich voor bij < 0,2% van de proefpersonen en deze waren evenwichtig verspreid over dapagliflozine 10 mg en placebo (zie rubriek 4.4).
In de DECLARE-studie was het aantal patiënten met voorvallen die zouden kunnen wijzen op volumedepletie gebalanceerd tussen de behandelgroepen: 213 (2,5%) en 207 (2,4%) in respectievelijk de dapagliflozine- en placebogroep. Ernstige bijwerkingen werden gerapporteerd bij 81 (0,9%) en 70 (0,8%) van de patiënten in respectievelijk de dapagliflozine- en placebogroep. De voorvallen waren over het algemeen gebalanceerd tussen de behandelgroepen over subgroepen van leeftijd, gebruik van diuretica, bloeddruk en gebruik van angiotensineconverterend-enzymremmers (ACE-remmers)/type1-angiotensine-II-receptorblokkers (ARB’s). Bij patiënten met een eGFR < 60 ml/min/1,73 m2 op baseline, waren er 19 voorvallen van ernstige bijwerkingen die zouden kunnen wijzen op volumedepletie in de dapagliflozinegroep en 13 voorvallen in de placebogroep.
In de DAPA-HF-studie was het aantal patiënten met voorvallen die zouden kunnen wijzen op volumedepletie 170 (7,2%) in de dapagliflozinegroep en 153 (6,5%) in de placebogroep. Er waren minder patiënten met ernstige voorvallen van symptomen die zouden kunnen wijzen op volumedepletie in de dapagliflozinegroep (23 [1,0%]) in vergelijking met de placebogroep (38 [1,6%]). De resultaten waren vergelijkbaar ongeacht de aanwezigheid van diabetes bij baseline en de baseline-eGFR. In de DELIVER-studie was het aantal patiënten met ernstige voorvallen van symptomen die zouden kunnen wijzen op volumedepletie 35 (1,1%) in de dapagliflozinegroep en 31 (1,0%) in de placebogroep.
In de DAPA-CKD-studie was het aantal patiënten met voorvallen die zouden kunnen wijzen op volumedepletie 120 (5,6%) in de dapagliflozinegroep en 84 (3,9%) in de placebogroep. Er waren 16 (0,7%) patiënten met ernstige voorvallen van symptomen die zouden kunnen wijzen op volumedepletie in de dapagliflozinegroep en 15 (0,7%) patiënten in de placebogroep.
Diabetische ketoacidose bij diabetes mellitus type 2
In de DECLARE-studie, met een mediane blootstelling van 48 maanden, werden voorvallen van DKA gerapporteerd bij 27 patiënten in de groep met dapagliflozine 10 mg en bij 12 patiënten in de placebogroep. De voorvallen waren gelijkmatig verdeeld over de studieperiode. Van de 27 patiënten met DKA-voorvallen in de dapagliflozinegroep, ontvingen er 22 gelijktijdige insulinebehandeling op het moment van het voorval. Precipiterende factoren voor DKA waren zoals verwacht in een populatie met diabetes mellitus type 2 (zie rubriek 4.4).
In de DAPA-HF-studie werden voorvallen van DKA gemeld bij 3 patiënten met diabetes mellitus type 2 in de dapagliflozinegroep, en geen in de placebogroep. In de DELIVER-studie werden voorvallen van DKA gemeld bij 2 patiënten met diabetes mellitus type 2 in de dapagliflozinegroep en bij geen enkele in de placebogroep.
In de DAPA-CKD-studie werden bij geen enkele patiënt in de dapagliflozinegroep voorvallen van DKA gemeld en bij 2 patiënten met diabetes mellitus type 2 in de placebogroep.
Urineweginfecties
In de gepoolde veiligheidsdata van 13 studies werden urineweginfecties vaker gerapporteerd voor dapagliflozine 10 mg dan voor placebo (respectievelijk 4,7% versus 3,5%; zie rubriek 4.4). De meeste gevallen van infectie waren mild tot matig en proefpersonen reageerden goed op een initiële kuur van de standaardbehandeling; de infecties leidden zelden tot staken van de dapagliflozinebehandeling. Deze infecties werden vaker gemeld bij vrouwen en proefpersonen met een voorgeschiedenis hadden een grotere kans om een terugkerende infectie te krijgen.
In de DECLARE-studie werden ernstige voorvallen van urineweginfecties minder vaak gerapporteerd voor dapagliflozine 10 mg vergeleken met placebo: 79 (0,9%) voorvallen versus 109 (1,3%) voorvallen, respectievelijk.
In de DAPA-HF-studie was het aantal patiënten met ernstige bijwerkingen van urineweginfecties 14 (0,6%) in de dapagliflozinegroep en 17 (0,7%) in de placebogroep. Er waren 5 (0,2%) patiënten met bijwerkingen die leidden tot stopzetting van de behandeling als gevolg van urineweginfecties in zowel de dapagliflozinegroep als in de placebogroep. In de DELIVER-studie was het aantal patiënten met ernstige bijwerkingen van urineweginfecties 41 (1,3%) in de dapagliflozinegroep en 37 (1,2%) in de placebogroep. Er waren 13 (0,4%) patiënten met bijwerkingen die leidden tot stopzetting van de behandeling als gevolg van urineweginfecties in de dapagliflozinegroep en 9 (0,3%) in de placebogroep.
In de DAPA-CKD-studie was het aantal patiënten met ernstige bijwerkingen van urineweginfecties 29 (1,3%) in de dapagliflozinegroep en 18 (0,8%) in de placebogroep. Er waren 8 (0,4%) patiënten met bijwerkingen die leidden tot stopzetting als gevolg van urineweginfecties in de dapagliflozinegroep en 3 (0,1%) patiënten in de placebogroep. De aantallen patiënten zonder diabetes die ernstige bijwerkingen van urineweginfecties of bijwerkingen die leidden tot stopzetting als gevolg van urineweginfecties meldden, waren vergelijkbaar tussen de behandelgroepen (6 [0,9%] versus 4 [0,6%] voor ernstige bijwerkingen en 1 [0,1%] versus 0 voor bijwerkingen die leidden tot stopzetting, respectievelijk in de dapagliflozine- en placebogroep).
Verhoogd creatinine
Bijwerkingen gerelateerd aan verhoogd creatinine zijn gegroepeerd (bv. verminderde nierklaring creatinine, nierfunctiestoornis, verhoogd bloedcreatinine en verminderde glomerulaire filtratiesnelheid). In de gepoolde veiligheidsdata van 13 studies is deze groepering van bijwerkingen gemeld bij resp. 3,2% en 1,8% van de patiënten die met dapagliflozine 10 mg en met placebo behandeld werden. Bij patiënten met een normale nierfunctie of een lichte nierfunctiestoornis (baseline eGFR ≥ 60 ml/min/1,73 m2) werd deze groepering van bijwerkingen gemeld bij resp. 1,3% en 0,8% van de patiënten die dapagliflozine 10 mg en placebo kregen. Deze bijwerkingen kwamen vaker voor bij patiënten met een baseline eGFR ≥ 30 en < 60 ml/min/1,73 m2 (18,5% bij dapagliflozine 10 mg versus 9.3% bij placebo).
Nadere evaluatie van de patiënten met bijwerkingen gerelateerd aan de nieren liet zien dat de meesten veranderingen in serumcreatinine hadden van ≤ 44 micromol/l (≤ 0,5 mg/dl) ten opzichte van baseline. De verhogingen in creatinine waren in het algemeen van voorbijgaande aard tijdens continue behandeling of reversibel na staken van de behandeling.
In de DECLARE-studie, waarin oudere patiënten en patiënten met nierinsufficiëntie (eGFR minder dan 60 ml/min/1,73 m2) waren opgenomen, nam de eGFR in de loop van de tijd in beide behandelgroepen af. Na 1 jaar was de gemiddelde eGFR iets lager, en na 4 jaar was de gemiddelde eGFR iets hoger in de dapagliflozinegroep vergeleken met de placebogroep.
In de DAPA-HF- en DELIVER-studies daalde de eGFR in de loop van de tijd in zowel de dapagliflozine- als in de placebogroep. In de DAPA-HF-studie was de initiële daling van de gemiddelde eGFR ‑4,3 ml/min/1,73 m2 in de dapagliflozinegroep en ‑1,1 ml/min/1,73 m2 in de placebogroep. Na 20 maanden was de verandering in eGFR ten opzichte van de baseline vergelijkbaar tussen de behandelingsgroepen: ‑5,3 ml/min/1,73 m2 voor dapagliflozine en ‑4,5 ml/min/1,73 m2 voor placebo. In de DELIVER-studie was de daling van de gemiddelde eGFR na 1 maand ‑3,7 ml/min/1,73 m2 in de dapagliflozinegroep en ‑0,4 ml/min/1,73 m2 in de placebogroep. Na 24 maanden was de verandering in eGFR ten opzichte van de baseline vergelijkbaar tussen de behandelgroepen: ‑4,2 ml/min/1,73 m2 in de dapagliflozinegroep en ‑3,2 ml/min/1,73m2 in de placebogroep.
In de DAPA-CKD-studie daalde de eGFR in de loop van de tijd in zowel de dapagliflozine- als in de placebogroep. De initiële daling (dag 14) van de gemiddelde eGFR was -4,0 ml/min/1,73 m2 in de dapagliflozinegroep en -0,8 ml/min/1,73 m2 in de placebogroep. Na 28 maanden was de verandering in eGFR ten opzichte van baseline -7,4 ml/min/1,73 m2 in de dapagliflozinegroep en -8,6 ml/min/1,73 m2 in de placebogroep.
Pediatrische patiënten
Het veiligheidsprofiel van dapagliflozine dat werd waargenomen in een klinische studie bij kinderen van 10 jaar en ouder met diabetes mellitus type 2 (zie rubriek 5.1) was vergelijkbaar met het profiel dat werd waargenomen in studies bij volwassenen.
Melden van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be


7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
AstraZeneca AB
SE-151 85 Södertälje
Zweden
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Forxiga 5 mg, filmomhulde tabletten
EU/1/12/795/001 14 filmomhulde tabletten
EU/1/12/795/002 28 filmomhulde tabletten
EU/1/12/795/003 98 filmomhulde tabletten
EU/1/12/795/004 30 x 1 (eenheidsdosis) filmomhulde tabletten
EU/1/12/795/005 90 x 1 (eenheidsdosis) filmomhulde tabletten
Forxiga 10 mg, filmomhulde tabletten
EU/1/12/795/006 14 filmomhulde tabletten
EU/1/12/795/007 28 filmomhulde tabletten
EU/1/12/795/008 98 filmomhulde tabletten
EU/1/12/795/009 30 x 1 (eenheidsdosis) filmomhulde tabletten
EU/1/12/795/010 90 x 1 (eenheidsdosis) filmomhulde tabletten
EU/1/12/795/011 10 x 1 (eenheidsdosis) filmomhulde tabletten
10. DATUM VAN HERZIENING VAN DE TEKST
03/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3018165 | FORXIGA 10 MG FILMOMHULDE COMP 98 X 10 MG | A10BK01 | € 144,17 | - | Ja | € 15,9 | € 10,5 |
| 3018173 | FORXIGA 10 MG FILMOMHULDE COMP 28 X 10 MG | A10BK01 | € 47,7 | - | Ja | € 2 | € 1 |