SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Trulicity 0,75 mg oplossing voor injectie in voorgevulde pen.
Trulicity 1,5 mg oplossing voor injectie in voorgevulde pen.
Trulicity 3 mg oplossing voor injectie in voorgevulde pen.
Trulicity 4,5 mg oplossing voor injectie in voorgevulde pen.
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Trulicity 0,75 mg oplossing voor injectie in voorgevulde pen
Elke voorgevulde pen bevat 0,75 mg dulaglutide* in 0,5 ml oplossing.
Hulpstof met bekend effect:
Eén ml oplossing bevat 0,20 mg polysorbaat 80.
Trulicity 1,5 mg oplossing voor injectie in voorgevulde pen
Elke voorgevulde pen bevat 1,5 mg dulaglutide* in 0,5 ml oplossing.
Hulpstof met bekend effect:
Eén ml oplossing bevat 0,20 mg polysorbaat 80.
Trulicity 3 mg oplossing voor injectie in voorgevulde pen
Elke voorgevulde pen bevat 3 mg dulaglutide* in 0,5 ml oplossing.
Hulpstof met bekend effect:
Eén ml oplossing bevat 0,25 mg polysorbaat 80.
Trulicity 4,5 mg oplossing voor injectie in voorgevulde pen
Elke voorgevulde pen bevat 4,5 mg dulaglutide* in 0,5 ml oplossing.
Hulpstof met bekend effect:
Eén ml oplossing bevat 0,25 mg polysorbaat 80.
*geproduceerd in CHO-cellen met DNA- recombinatietechniek.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie.
Heldere, kleurloze oplossing.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Type 2-diabetes mellitus
Trulicity is geïndiceerd voor de behandeling van patiënten van 10 jaar en ouder met onvoldoende gereguleerde diabetes mellitus type 2 als toevoeging aan dieet en lichaamsbeweging
- als monotherapie wanneer metformine ongeschikt wordt geacht als gevolg van intolerantie of contra-indicaties.
- in aanvulling op andere geneesmiddelen voor de behandeling van diabetes.
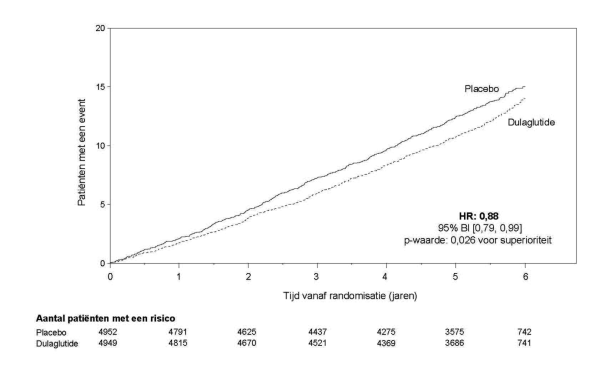
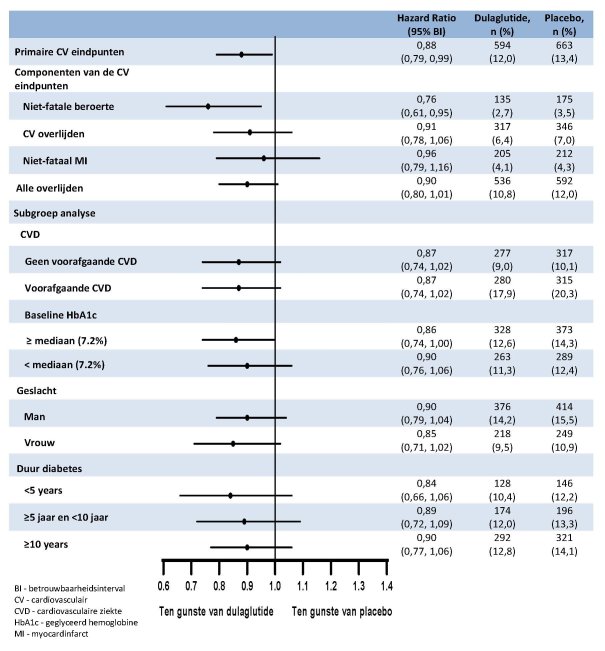
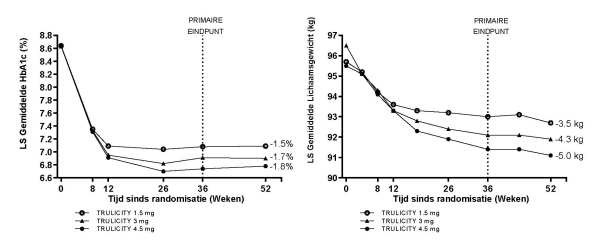
Voor onderzoeksresultaten met betrekking tot combinaties, effecten op glykemische controle en cardiovasculaire events, en de onderzochte populaties, zie rubriek 4.4, 4.5 en 5.1.
4.2 Dosering en wijze van toediening
Dosering
Volwassenen
Monotherapie
De aanbevolen dosering is 0,75 mg eenmaal per week.
Adjuvante therapie
De aanbevolen dosering is 1,5 mg eenmaal per week.
Indien nodig,
- kan de dosering van 1,5 mg na ten minste 4 weken worden verhoogd tot 3 mg eenmaal per week.
- kan de dosering van 3 mg na ten minste 4 weken worden verhoogd tot 4,5 mg eenmaal per week.
De maximale dosering is 4,5 mg eenmaal per week.
Pediatrische patiënten
De startdosering voor pediatrische patiënten van 10 jaar en ouder is 0,75 mg eenmaal per week.
Indien nodig, kan de dosering na minimaal 4 weken worden verhoogd tot 1,5 mg eenmaal per week. De maximale dosering is 1,5 mg eenmaal per week.
Combinatietherapie
Als Trulicity wordt toegevoegd aan een eerder ingestelde behandeling met metformine en/of pioglitazon, kan de huidige dosis metformine en/of pioglitazon worden voortgezet. Als Trulicity wordt toegevoegd aan een eerder ingestelde behandeling met metformine en/of een natriumglucose-cotransporter 2-remmer (SGLT2-remmer), kan de huidige dosis metformine en/of SGLT2-remmer worden voortgezet. Als het wordt toegevoegd aan een eerder ingestelde behandeling met een sulfonylureumderivaat of insuline, kan verlaging van de dosis van het sulfonylureumderivaat of de insuline worden overwogen om de kans op hypoglykemie te verminderen (zie rubriek 4.4 en 4.8).
Bij gebruik van Trulicity hoeven patiënten hun bloedglucose niet zelf te controleren. Zelfcontrole van de bloedglucose is nodig om de dosis van het sulfonylureumderivaat of de insuline aan te passen, met name wanneer de behandeling met Trulicity is gestart en de dosis insuline is verlaagd. Een stapsgewijze benadering van de verlaging van de insulinedosis wordt aanbevolen.
Gemiste doses
Als een dosis wordt vergeten, moet deze zo snel mogelijk worden toegediend als de tijd tot de volgende geplande dosis minstens 3 dagen (72 uur) is. Als de tijd tot de volgende geplande dosis minder dan 3 dagen (72 uur) is, moet de vergeten dosis worden overgeslagen en moet de volgende dosis op de geplande dag worden toegediend. In beide gevallen kunnen patiënten hun normale toedieningsschema van eenmaal per week hervatten.
Speciale patiëntengroepen
Ouderen
De dosis hoeft niet op basis van leeftijd te worden aangepast (zie rubriek 5.2).
Nierfunctiestoornis
Bij patiënten met lichte, matige of ernstige nierfunctiestoornis (eGFR <90 tot >15 ml/min/1,73 m2) hoeft de dosering niet te worden aangepast.
Er is zeer beperkte ervaring bij patiënten met nierziekte in het eindstadium (<15 ml/min/1,73 m2), daarom kan Trulicity bij deze populatie niet worden aanbevolen (zie rubriek 5.1 en 5.2).
Leverfunctiestoornis
Bij patiënten met leverfunctiestoornis hoeft de dosering niet te worden aangepast.
Pediatrische patiënten
De veiligheid en werkzaamheid van dulaglutide bij kinderen jonger dan 10 jaar zijn niet vastgesteld en er zijn geen gegevens beschikbaar (zie rubriek 5.1 en 5.2).
Wijze van toediening
Trulicity dient subcutaan in de buik, dij of bovenarm te worden toegediend. Het mag niet intraveneus of intramusculair worden toegediend.
De dosis kan op elk moment op de dag worden toegediend, met of zonder voedsel.
Zo nodig kan de dag van de wekelijkse toediening worden veranderd, zolang de vorige dosis 3 of meer dagen (72 uur) eerder is toegediend.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof(fen) of voor een van de in rubriek 6.1 vermelde hulpstof(fen).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
In de afgeronde initiële fase 2- en fase 3-registratiestudies ter onderbouwing van dulaglutide 0,75 mg en 1,5 mg zijn 4.006 patiënten blootgesteld aan dulaglutide alleen of in combinatie met andere glucoseverlagende geneesmiddelen. De meest frequent gemelde bijwerkingen in klinische studies waren gastro-intestinaal, waaronder misselijkheid, braken en diarree. Over het algemeen waren deze reacties licht of matig en van voorbijgaande aard. Resultaten uit de studie naar cardiovasculaire uitkomsten op lange termijn met 4949 naar dulaglutide gerandomiseerde patiënten die werden gevolgd gedurende een periode met een mediaan van 5,4 jaar, waren consistent met deze bevindingen.
Tabel met overzicht van bijwerkingen
De volgende bijwerkingen zijn vastgesteld op basis van beoordeling van de volledige duur van de klinische fase 2- en fase 3-studies, de studie naar cardiovasculaire uitkomsten op lange termijn en van post-marketing meldingen. De bijwerkingen staan in tabel 1 als MedDRA-geprefereerde term per systeem/orgaanklasse en in volgorde van afnemende incidentie (zeer vaak: ≥ 1/10; vaak: ≥ 1/100, < 1/10; soms: ≥ 1/1.000, < 1/100; zelden: ≥ 1/10.000, < 1/1.000; zeer zelden: < 1/10.000 en niet bekend: kan met de beschikbare gegevens niet worden bepaald). Binnen elke incidentiegroep staan de bijwerkingen in volgorde van afnemende frequentie. Frequenties van voorvallen zijn berekend op basis van hun incidentie in de fase 2- en fase 3-registratiestudies.
Tabel 1: Frequentie van bijwerkingen van dulaglutide
Systeem/ orgaanklasse | Zeer vaak | Vaak | Soms | Zelden | Niet bekend |
Immuunsysteem-aandoeningen |
|
| Overgevoelig-heid | Anafylacti-sche reactie# |
|
Voedings- en stof-wisselingsstoor-nissen | Hypoglykemie* (bij gebruik in combinatie met insuline, | Hypoglykemie* (bij gebruik als monotherapie of in combinatie met metformine plus pioglitazon) | Dehydratie |
|
|
Zenuwstelsel-aandoeningen |
|
| Dysgeusie |
|
|
Maagdarmstelsel-aandoeningen | Misselijkheid, diarree, | Verminderde eetlust, dyspepsie, obstipatie, flatulentie, opgezette buik, gastro-oesofageale- refluxziekte, eructatie |
| Acute pancreatitis, | Niet-mecha-nische darm-obstructie |
Lever- en galaandoeningen |
|
| Cholelithiasis, cholecystitis |
|
|
Huid- en onderhuidaan-doeningen |
|
|
| Angio-oedeem# |
|
Algemene aandoe-ningen en toedie-ningsplaatsstoor-nissen |
| Vermoeidheid | Reacties op de injectieplaats$ |
|
|
Onderzoeken |
| Sinustachycardie, eerstegraads atrio-ventriculair blok (AVB) |
|
|
|
# Uit post-marketing meldingen
* Gedocumenteerde, symptomatische hypoglykemie met bloedglucose ≤ 3,9 mmol/l
† Met dulaglutide 0,75 mg traden de bijwerkingen op in de frequentie van de eerstvolgende lagere incidentiegroep.
$ De frequentie die werd waargenomen in een pediatrisch onderzoek was vaak; 3,9% (2 patiënten) in de groep met 0,75 mg dulaglutide, 3,8% (2 patiënten) in de groep met 1,5 mg dulaglutide en 2% (1 patiënt) in de placebogroep. Alle voorvallen waren licht tot matig van ernst.
Omschrijving van geselecteerde bijwerkingen
Hypoglykemie
Als dulaglutide 0,75 mg en 1,5 mg werden gebruikt als monotherapie of in combinatie met metformine alleen of metformine en pioglitazon, waren de incidenties van gedocumenteerde symptomatische hypoglykemie 5,9% tot 10,9% en de frequenties waren 0,14 tot 0,62 voorvallen/patiënt/jaar; er zijn geen episodes van ernstige hypoglykemie gemeld.
De incidenties van gedocumenteerde symptomatische hypoglykemie als respectievelijk 0,75 mg en 1,5 mg dulaglutide werden gebruikt in combinatie met een sulfonylureumderivaat en metformine waren 39,0% en 40,3% en de frequenties waren 1,67 en 1,67 voorvallen/patiënt/jaar. De incidenties van ernstige hypoglykemie waren 0% en 0,7%, en de frequenties waren 0,00 en 0,01 voorvallen/patiënt/jaar respectievelijk voor elke dosis. De incidentie van gedocumenteerde symptomatische hypoglykemie wanneer 1,5 mg dulaglutide werd gebruikt met alleen een sulfonylureumderivaat was 11,3% en de frequentie was 0,90 voorvallen/patiënt/jaar, en er waren geen gevallen van ernstige hypoglykemie.
De incidentie van gedocumenteerde symptomatische hypoglykemie wanneer dulaglutide 1,5 mg werd gebruikt in combinatie met insuline glargine was 35,3% en de frequentie was 3,38 voorvallen/patiënt/jaar. De incidentie van ernstige hypoglykemie was 0,7% en de frequentie was 0,01 voorval/patiënt/jaar.
De incidenties wanneer respectievelijk 0,75 mg en 1,5 mg dulaglutide werden gebruikt in combinatie met prandiale insuline waren 85,3% en 80,0% en de frequenties waren 35,66 en 31,06 voorvallen/patiënt/jaar. De incidenties van ernstige hypoglykemie waren 2,4% en 3,4%, en de frequenties waren 0,05 en 0,06 voorvallen/patiënt/jaar.
In een fase 3-studie waarin dulaglutide 1,5 mg, 3 mg en 4,5 mg in combinatie met metformine werden gebruikt, waren t/m week 52 de incidenties van gedocumenteerde symptomatische hypoglykemie respectievelijk 3,1%, 2,4% en 3,1% en waren de aantallen 0,07, 0,05 en 0,07 voorvallen/patiënt/ jaar; er werd 1 episode van ernstige hypoglykemie gemeld bij achtereenvolgens dulaglutide 1,5 mg en 4,5 mg.
Bijwerkingen aan het maagdarmstelsel
Cumulatieve melding van bijwerkingen aan het maagdarmstelsel tot 104 weken met respectievelijk 0,75 mg en 1,5 mg dulaglutide waren misselijkheid (12,9% en 21,2%), diarree (10,7% en 13,7%) en braken (6,9% en 11,5%). Deze waren over het algemeen licht of matig ernstig met een gemelde piek tijdens de eerste 2 weken van behandeling gevolgd door een snelle afname gedurende de daaropvolgende 4 weken, waarna de frequentie relatief constant bleef.
In een fase 3-studie met doseringen dulaglutide van respectievelijk 1,5 mg, 3 mg en 4,5 mg werden tot en met week 52 misselijkheid (14,2%, 16,1% en 17,3%), diarree (7,7%, 12,0% en 11,6%) en braken (6,4%, 9,1% en 10,1%) opgenomen in de cumulatieve rapportage van gastro-intestinale voorvallen.
In klinisch-farmacologische, tot 6 weken durende studies bij patiënten met diabetes mellitus type 2 werden de meeste bijwerkingen aan het maagdarmstelsel gemeld tijdens de eerste 2-3 dagen na de aanvangsdosis en namen deze bij volgende doses af.
Acute pancreatitis
De incidentie van acute pancreatitis in fase 2- en 3-registratiestudies was 0,07% voor dulaglutide tegen 0,14% voor placebo en 0,19% voor comparators met of zonder additionele antidiabetische achtergrondtherapie. Acute pancreatitis en pancreatitis zijn ook gemeld na het in de handel brengen.
Pancreasenzymen
Dulaglutide gaat gepaard met gemiddelde toenames ten opzichte van de uitgangswaarde van de pancreasenzymen (lipase en/of pancreatisch amylase) van 11% tot 21% (zie rubriek 4.4). In afwezigheid van andere klachten en symptomen van acute pancreatitis zijn verhogingen van de pancreasenzymen alleen niet voorspellend voor acute pancreatitis.
Verhoogde hartslag
Met respectievelijk 0,75 mg en 1,5 mg dulaglutide zijn geringe gemiddelde verhogingen van de hartslag van 2 tot 4 slagen per minuut (bpm) en een incidentie van 1,3% en 1,4% van sinustachycardie met een gelijktijdige verhoging t.o.v. de uitgangswaarde ≥ 15 bpm waargenomen.
In een fase 3-studie met doseringen dulaglutide van 1,5 mg, 3 mg en 4,5 mg was de incidentie sinustachycardie, met een gelijktijdige toename van de hartslag met ≥ 15 bpm vanaf de baseline, respectievelijk 2,6%, 1,9% en 2,6%. Er werden gemiddelde toenames in hartslag waargenomen van 1-4 slagen per minuut (bpm).
Eerstegraads AV-blok/verlengd PR-interval
Met respectievelijk 0,75 mg en 1,5 mg dulaglutide zijn geringe gemiddelde verhogingen t.o.v. de uitgangswaarde van het PR-interval van 2 tot 3 msec en een incidentie van 1,5% en 2,4% van eerstegraads AV-blok waargenomen.
In een fase 3-studie met doseringen dulaglutide van 1,5 mg, 3 mg en 4,5 mg was de incidentie van eerstegraads AV-blok respectievelijk 1,2%, 3,8% en 1,7%. Er werden vanaf de baseline gemiddelde toenames in het PR-interval waargenomen van 3-5 msec.
Immunogeniciteit
In registratiestudies ging behandeling met dulaglutide gepaard met een incidentie van 1,6% van tijdens de behandeling gevormde antistoffen tegen dulaglutide, wat erop wijst dat de structurele aanpassingen in de GLP-1- en gemodificeerde IgG4-delen van de dulaglutidemolecuul, samen met een hoge homologie met natief GLP-1 en natief IgG4, de kans op een immuunrespons tegen dulaglutide tot een minimum beperken. Patiënten met antistoffen tegen dulaglutide hadden over het algemeen een lage titer en hoewel het aantal patiënten met antistoffen tegen dulaglutide laag was, werd uit bestudering van de fase 3-gegevens geen duidelijke invloed zichtbaar van antistoffen tegen dulaglutide op veranderingen in HbA1c. Geen van de patiënten met systemische overgevoeligheid ontwikkelde antilichamen tegen dulaglutide.
Overgevoeligheid
In de fase 2- en fase 3-registratiestudies is systemische overgevoeligheid (bijvoorbeeld urticaria, oedeem) gemeld bij 0,5% van de patiënten die dulaglutide kregen. Gevallen van anafylactische reactie zijn zelden gemeld bij gebruik van in de handel gebrachte dulaglutide.
Reacties op de injectieplaats
Bijwerkingen op de injectieplaats zijn gemeld bij 1,9% van de patiënten die dulaglutide kregen. Mogelijk immuungemedieerde bijwerkingen op de injectieplaats (zoals uitslag, erytheem) zijn gemeld bij 0,7% van de patiënten en waren over het algemeen licht.
Stopzetting wegens een bijwerking
In studies die 26 weken duurden, was de incidentie van stopzetting wegens bijwerkingen 2,6% (0,75 mg) en 6,1% (1,5 mg) voor dulaglutide versus 3,7% voor placebo. Gedurende het gehele onderzoek (tot 104 weken) was de incidentie van stopzetting wegens bijwerkingen 5,1% (0,75 mg) en 8,4% (1,5 mg) voor dulaglutide. De meest voorkomende bijwerkingen die tot stopzetting leidden van respectievelijk 0,75 mg en 1,5 mg dulaglutide waren misselijkheid (1,0%, 1,9%), diarree (0,5%, 0,6%) en braken (0,4%, 0,6%), en werden over het algemeen binnen de eerste 4-6 weken gemeld.
In een fase 3-studie met doseringen dulaglutide van 1,5 mg, 3 mg en 4,5 mg was de incidentie van stopzetting wegens bijwerkingen na 52 weken 6,0% (1,5 mg), 7,0% (3 mg) en 8,5% (4,5 mg). De meest frequente bijwerkingen die leidden tot discontinuering van respectievelijk 1,5 mg, 3 mg en 4,5 mg waren misselijkheid (1,3%, 1,3%, 1,5%), diarree (0,2%, 1,0%, 1,0%) en braken (0,0%, 0,8%, 1,3%).
Dulaglutidedoseringen van 3 mg en 4,5 mg
Het veiligheidsprofiel bij patiënten, behandeld met eenmaal per week 3 mg en 4,5 mg dulaglutide, is consistent met het hierboven beschreven veiligheidsprofiel voor doseringen dulaglutide van 0,75 mg en 1,5 mg eenmaal per week.
Pediatrische patiënten
Het veiligheidsprofiel bij pediatrische patiënten van 10 jaar en ouder die eenmaal per week werden behandeld met 0,75 mg en 1,5 mg dulaglutide is vergelijkbaar met het veiligheidsprofiel dat hierboven is beschreven voor volwassen patiënten.
Het immunogeniciteitsprofiel bij pediatrische patiënten die met dulaglutide werden behandeld, komt overeen met het hierboven beschreven profiel voor volwassen patiënten. 2,1% en 4,0% van de patiënten die in het pediatrische onderzoek werden behandeld met respectievelijk placebo en dulaglutide ontwikkelden tijdens de behandeling gevormde antistoffen tegen dulaglutide.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten, www.fagg.be, Afdeling Vigilantie: Website: www.eenbijwerkingmelden.be, e-mail: adr@fagg-afmps.be.
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Eli Lilly Nederland B.V., Orteliuslaan 1000, 3528 BD Utrecht, Nederland.
8. NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/14/956/001
EU/1/14/956/002
EU/1/14/956/003
EU/1/14/956/006
EU/1/14/956/007
EU/1/14/956/008
EU/1/14/956/011
EU/1/14/956/012
EU/1/14/956/013
EU/1/14/956/014
EU/1/14/956/015
EU/1/14/956/016
10. DATUM VAN HERZIENING VAN DE TEKST 01/2026
AFLEVERINGSWIJZE Geneesmiddel op medisch voorschrift.
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3275971 | TRULICITY 0,75MG/0,5ML OPL INJ VOORGEVULDE PEN 4 | A10BJ05 | € 104,05 | - | Ja | € 2 | € 1 |
| 3275989 | TRULICITY 1,50MG/0,5ML OPL INJ VOORGEVULDE PEN 4 | A10BJ05 | € 104,05 | - | Ja | € 2 | € 1 |