1. NAAM VAN HET GENEESMIDDEL
XGEVA 120 mg oplossing voor injectie
XGEVA 120 mg oplossing voor injectie in voorgevulde spuit
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke injectieflacon bevat 120 mg denosumab in 1,7 ml oplossing (70 mg/ml).
Elke voorgevulde spuit bevat 120 mg denosumab in 1,0 ml oplossing (120 mg/ml).
Denosumab is een humaan monoklonaal IgG2‑antilichaam geproduceerd in een zoogdiercellijn (Chinese hamster ovariumcellen) via recombinant DNA‑technologie.
Hulpstof met bekend effect
Elke 1,7 ml oplossing bevat 78 mg sorbitol (E420).
Elke 1,0 ml oplossing bevat 37 mg sorbitol (E420) en 6,1 mg L‑fenylalanine.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
XGEVA 120 mg oplossing voor injectie
Oplossing voor injectie (injectievloeistof).
XGEVA 120 mg oplossing voor injectie in voorgevulde spuit
Oplossing voor injectie (injectievloeistof).
Heldere, kleurloze tot lichtgele oplossing die sporen kan bevatten van doorschijnende tot witte eiwitachtige deeltjes.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Preventie van botcomplicaties (pathologische fractuur, bestraling van bot, ruggenmergcompressie of chirurgie van het bot) bij volwassenen met gevorderde maligniteiten waarbij bot is betrokken (zie rubriek 5.1).
Behandeling van volwassenen en adolescenten met een volgroeid skelet met reusceltumor van het bot (‘giant cell tumour of bone’) die niet‑reseceerbaar is of waarbij chirurgische resectie waarschijnlijk leidt tot ernstige morbiditeit.
4.2 Dosering en wijze van toediening
XGEVA moet worden toegediend onder de verantwoordelijkheid van een beroepsbeoefenaar in de gezondheidszorg.
Dosering
Dagelijkse suppletie met ten minste 500 mg calcium en 400 IE vitamine D is bij alle patiënten noodzakelijk, tenzij er sprake is van hypercalciëmie (zie rubriek 4.4).
De bijsluiter en de herinneringskaart voor patiënten moeten worden meegegeven aan patiënten die worden behandeld met XGEVA.
Preventie van botcomplicaties bij volwassenen met gevorderde maligniteiten waarbij bot is betrokken
De aanbevolen dosering is 120 mg toegediend als een enkelvoudige subcutane injectie eenmaal per 4 weken in dij, buik of bovenarm.
Reusceltumor van het bot
De aanbevolen dosering van XGEVA is 120 mg toegediend als een enkelvoudige subcutane injectie eenmaal per 4 weken in de dij, buik of bovenarm met extra doses van 120 mg op dag 8 en 15 van de behandeling in de eerste maand van de therapie.
Patiënten in de fase II‑studie die een volledige resectie van een reusceltumor van het bot hebben ondergaan, zijn na de chirurgische ingreep nog eens 6 maanden behandeld, overeenkomstig het studie protocol.
Patiënten met een reusceltumor van het bot dienen met regelmatige intervallen te worden onderzocht om na te gaan of ze nog steeds baat hebben bij de behandeling. Bij patiënten bij wie de ziekte met XGEVA onder controle is gebracht, is het effect van het onderbreken of stopzetten van de behandeling niet beoordeeld. Beperkte gegevens bij deze patiënten duiden echter niet op een terugval na stopzetting van de behandeling.
Nierfunctiestoornis
Voor patiënten met een nierfunctiestoornis is geen dosisaanpassing nodig (zie rubriek 4.4 voor aanbevelingen voor de monitoring van calcium, en rubriek 4.8 en 5.2).
Leverfunctiestoornis
De veiligheid en werkzaamheid van denosumab zijn niet onderzocht bij patiënten met een leverfunctiestoornis (zie rubriek 5.2).
Ouderen (leeftijd ≥ 65)
Voor ouderen is geen dosisaanpassing nodig (zie rubriek 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van XGEVA zijn niet vastgesteld bij pediatrische patiënten (leeftijd < 18 jaar) behalve bij adolescenten met een volgroeid skelet (leeftijd 12‑17 jaar) met een reusceltumor van het bot.
XGEVA wordt niet aanbevolen bij pediatrische patiënten (leeftijd < 18 jaar) behalve bij adolescenten met een volgroeid skelet (leeftijd 12‑17 jaar) met een reusceltumor van het bot (zie rubriek 4.4).
Behandeling van adolescenten met een volgroeid skelet met een reusceltumor van het bot die niet‑reseceerbaar is of waarbij chirurgische resectie waarschijnlijk leidt tot ernstige morbiditeit: de dosering is gelijk aan die bij volwassenen.
Remming van RANK/RANK‑ligand (RANKL) werd in experimenteel onderzoek bij dieren geassocieerd met remming van botgroei en afwezigheid van tanddoorbraak. Deze veranderingen waren gedeeltelijk reversibel bij stopzetting van RANKL‑remming (zie rubriek 5.3).
Wijze van toediening
Voor subcutaan gebruik.
De XGEVA 120 mg/1,7 ml oplossing in een injectieflacon voor eenmalig gebruik:
De inhoud van de 120 mg/1,7 ml injectieflacon mag uitsluitend worden toegediend door een beroepsbeoefenaar in de gezondheidszorg.
De XGEVA 120 mg/1,0 ml oplossing in een voorgevulde spuit:
De inhoud van de 120 mg/1,0 ml voorgevulde spuit kan worden toegediend door een patiënt of verzorger die door een beroepsbeoefenaar in de gezondheidszorg is getraind in injectietechnieken. De eerste zelftoediening met de XGEVA voorgevulde spuit dient plaats te vinden onder toezicht van een beroepsbeoefenaar in de gezondheidszorg.
Voor instructies over het gebruik, de hantering en de verwijdering, zie rubriek 6.6.
4.3 Contra-indicaties
Overgevoeligheid voor het werkzame bestanddeel of voor één van de in rubriek 6.1 vermelde hulpstoffen.
Ernstige, onbehandelde hypocalciëmie (zie rubriek 4.4).
Niet‑genezen laesies als gevolg van kaak‑ of mondchirurgie.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Het algehele veiligheidsprofiel komt overeen voor alle goedgekeurde indicaties van XGEVA.
Hypocalciëmie is zeer vaak gemeld na toediening van XGEVA, vooral in de eerste twee weken. Hypocalciëmie kan ernstig en symptomatisch zijn (zie rubriek 4.8 – beschrijving van geselecteerde bijwerkingen). De afname van de serumcalciumspiegel werd over het algemeen adequaat opgevangen door calcium‑ en vitamine‑D‑suppletie. De meest voorkomende bijwerking met XGEVA is skeletspierstelselpijn. Gevallen van osteonecrose van de kaak (zie rubriek 4.4 en 4.8 – beschrijving van geselecteerde bijwerkingen) zijn vaak gezien bij patiënten die behandeld worden met XGEVA.
Tabel met bijwerkingen
De volgende conventie is gebruikt voor de classificatie van de bijwerkingen op basis van incidentiecijfers in vier klinische fase III‑onderzoeken, twee klinische fase II‑onderzoeken en postmarketingervaring (zie tabel 1): zeer vaak (≥ 1/10), vaak (≥ 1/100, < 1/10), soms (≥ 1/1.000, < 1/100), zelden (≥ 1/10.000, < 1/1.000), zeer zelden (< 1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen iedere frequentiegroep en systeem/orgaanklasse worden bijwerkingen gerangschikt naar afnemende ernst.
Tabel 1. Bijwerkingen die zijn gemeld bij patiënten met gevorderde maligniteiten waarbij bot was betrokken, multipel myeloom, of met reusceltumor van het bot
MedDRA systeem/orgaanklasse | Frequentiecategorie | Bijwerkingen |
Neoplasmata, benigne, maligne en niet-gespecificeerd (inclusief cysten en poliepen) | Vaak | Nieuwe primaire maligniteit1 |
Immuunsysteemaandoeningen | Zelden | Overgevoeligheid voor het geneesmiddel1 |
Zelden | Anafylactische reactie1 | |
Voedings- en stofwisselingsstoornissen | Zeer vaak | Hypocalciëmie1, 2 |
Vaak | Hypofosfatemie | |
Soms | Hypercalciëmie na het stopzetten van de behandeling bij patiënten met reusceltumor van het bot3 | |
Ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen | Zeer vaak | Dyspneu |
Maagdarmstelselaandoeningen | Zeer vaak | Diarree |
Vaak | Tandextractie | |
Huid- en onderhuidaandoeningen | Vaak | Hyperhidrosis |
Soms | Lichenoïde reacties door medicijngebruik1 | |
Skeletspierstelsel- en bindweefselaandoeningen | Zeer vaak | Skeletspierstelselpijn1 |
Vaak | Osteonecrose van de kaak1 | |
Soms | Atypische femurfractuur1 | |
Niet bekend | Osteonecrose van de uitwendige gehoorgang3,4 |
1 Zie rubriek Beschrijving van geselecteerde bijwerkingen
2 Zie rubriek Andere speciale patiëntengroepen
3 Zie rubriek 4.4
4 Klasse effect
Beschrijving van geselecteerde bijwerkingen
Hypocalciëmie
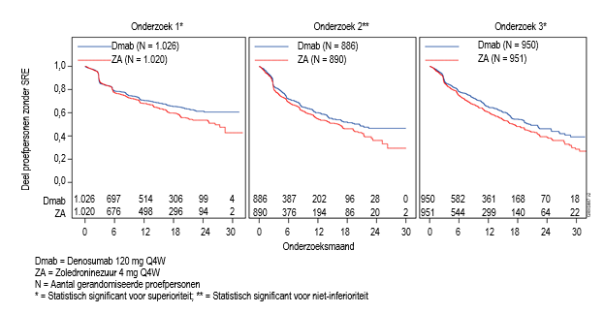
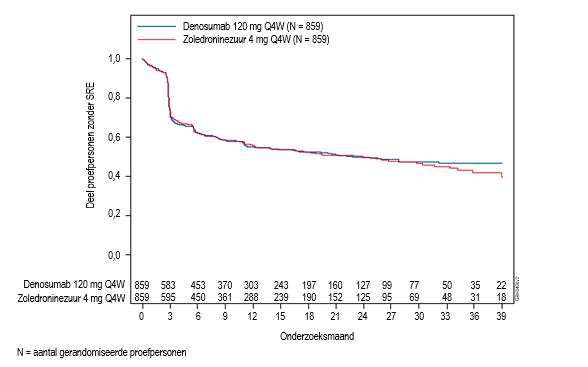
Een hogere incidentie van hypocalciëmie bij patiënten behandeld met denosumab in vergelijking met zoledroninezuur is waargenomen in klinische onderzoeken naar SRE‑preventie.
De hoogste incidentie van hypocalciëmie werd waargenomen in een fase III‑onderzoek bij patiënten met multipel myeloom. Hypocalciëmie werd gemeld bij 16,9% van de patiënten behandeld met XGEVA en bij 12,4% van de patiënten behandeld met zoledroninezuur. Een graad 3‑afname in serumcalciumspiegels werd waargenomen bij 1,4% van de patiënten die werden behandeld met XGEVA en bij 0,6% van de patiënten die werden behandeld met zoledroninezuur. Een graad 4‑afname in serumcalciumspiegels werd waargenomen bij 0,4% van de patiënten behandeld met XGEVA en bij 0,1% van de patiënten behandeld met zoledroninezuur.
In drie actief gecontroleerde klinische fase III‑onderzoeken bij patiënten met gevorderde maligniteiten waarbij bot was betrokken, werd bij 9,6% van de met XGEVA behandelde patiënten en bij 5,0% van de met zoledroninezuur behandelde patiënten hypocalciëmie gemeld.
Er deed zich een graad 3‑afname van de serumcalciumspiegel voor bij 2,5% van de met XGEVA behandelde patiënten en bij 1,2% van de patiënten behandeld met zoledroninezuur. Er was sprake van een graad 4‑afname van de serumcalciumspiegel bij 0,6% van de met XGEVA behandelde patiënten en bij 0,2% van de patiënten behandeld met zoledroninezuur (zie rubriek 4.4).
In twee klinische fase II‑onderzoeken met één onderzoeksarm bij patiënten met reusceltumor van het bot, werd bij 5,7% van de patiënten hypocalciëmie gemeld. Geen van de ongewenste voorvallen werden als ernstig beoordeeld.
In de postmarketingsetting is ernstige symptomatische hypocalciëmie (inclusief gevallen met fatale afloop) gemeld, waarbij de meeste gevallen optraden tijdens de eerste weken na het starten van de behandeling. Voorbeelden van klinische manifestaties van ernstige symptomatische hypocalciëmie omvatten onder meer verlenging van het QT‑interval, tetanie, epileptische aanvallen en veranderde mentale toestand (waaronder coma) (zie rubriek 4.4). Symptomen van hypocalciëmie in klinische onderzoeken omvatten paresthesieën of spierstijfheid, spiertrekkingen, spasmen en spierkrampen.
Osteonecrose van de kaak (ONJ)
In klinische studies was de incidentie van ONJ hoger naarmate men langer aan de therapie werd blootgesteld; ONJ werd ook vastgesteld na het stoppen van de behandeling met XGEVA, waarvan de meeste gevallen zich binnen 5 maanden na de laatste dosis voordeden. Patiënten met een voorgeschiedenis van ONJ of osteomyelitis van de kaak, een actieve aandoening van het gebit of de kaak waarbij een chirurgische ingreep noodzakelijk is, geen genezing na tandheelkundige behandeling/mondchirurgie of een geplande invasieve tandheelkundige ingreep werden uitgesloten van deelname aan de klinische onderzoeken.
Een hogere incidentie van ONJ bij patiënten die werden behandeld met denosumab vergeleken met zoledroninezuur werd waargenomen in klinische onderzoeken naar SRE‑preventie. De hoogste incidentie van ONJ werd waargenomen in een fase III‑onderzoek bij patiënten met multipel myeloom. In de dubbelblinde behandelfase van dit onderzoek werd ONJ bevestigd bij 5,9% van de met XGEVA behandelde patiënten (mediane blootstelling van 19,4 maanden, spreiding: 1‑52) en bij 3,2% van de patiënten behandeld met zoledroninezuur. Na voltooiing van de dubbelblinde behandelfase van dit onderzoek, was de incidentie van ONJ gecorrigeerd voor patiëntjaren van bevestigde ONJ in de XGEVA‑groep (mediane blootstelling van 19,4 maanden, spreiding: 1‑52), 2,0 per 100 patiëntjaren tijdens het eerste jaar van de behandeling, 5,0 in het tweede jaar en daarna 4,5. De mediane tijd tot ONJ was 18,7 maanden (spreiding: 1‑44).
In de primaire behandelfases van drie actief gecontroleerde klinische fase III‑onderzoeken bij patiënten met gevorderde maligniteiten waarbij bot was betrokken, werd ONJ bevestigd bij 1,8% van de met XGEVA behandelde patiënten (mediane blootstelling van 12,0 maanden; spreiding: 0,1‑40,5) en bij 1,3% van de met zoledroninezuur behandelde patiënten. De klinische kenmerken van deze gevallen waren in de behandelgroepen vergelijkbaar. Van de patiënten met bevestigde ONJ hadden de meesten (81% in beide behandelgroepen) een voorgeschiedenis van tandextractie, slechte mondhygiëne en/of gebruik van een tandheelkundig hulpmiddel. De meeste patiënten werden behandeld met chemotherapie.
De onderzoeken bij patiënten met borst- of prostaatkanker waren inclusief een verlengingsfase van de behandeling met XGEVA (mediane totale blootstelling van 14,9 maanden; spreiding: 0,1‑67,2). Tijdens de verlengingsfase van de behandeling werd ONJ bevestigd bij 6,9% van de patiënten met borstkanker en met prostaatkanker.
De totale incidentie van ONJ gecorrigeerd voor patiëntjaren was 1,1 per 100 patiëntjaren gedurende het eerste jaar van de behandeling, 3,7 in het tweede jaar en vervolgens 4,6. De mediane tijd tot ONJ was 20,6 maanden (spreiding: 4–53).
Een niet‑gerandomiseerd, retrospectief, observationeel onderzoek bij 2.877 patiënten met kanker die werden behandeld met XGEVA of zoledroninezuur in Zweden, Denemarken en Noorwegen toont aan dat het incidentiepercentage van medisch bevestigd ONJ na 5 jaar 5,7% was (95% BI: 4,4; 7,3; mediane follow‑uptijd van 20 maanden [bereik 0,2‑60]) in een cohort van patiënten die werden behandeld met XGEVA en 1,4% (95% BI: 0,8; 2,3; mediane follow‑uptijd van 13 maanden [bereik 0,1‑60]) in een apart cohort van patiënten die werden behandeld met zoledroninezuur. Het incidentiepercentage van ONJ na 5 jaar bij patiënten die van zoledroninezuur overstapten op XGEVA was 6,6% (95% BI: 4,2; 10,0; mediane follow‑uptijd van 13 maanden [bereik 0,2‑60]).
In een fase III onderzoek bij patiënten met niet‑uitgezaaide prostaatkanker (een patiëntenpopulatie waarvoor XGEVA niet is geïndiceerd), die langer, tot 7 jaar, zijn blootgesteld aan behandeling, was de incidentie van bevestigde ONJ gecorrigeerd voor patiëntjaren 1,1 per 100 patiëntjaren gedurende het eerste jaar van de behandeling, 3,0 in het tweede jaar, en vervolgens 7,1.
In een langdurig, open‑label klinisch fase II-onderzoek bij patiënten met reusceltumor van het bot (studie 6, zie rubriek 5.1) werd ONJ bevestigd bij 6,8% van de patiënten, waaronder één adolescent (mediaan aantal doseringen was 34; spreiding 4‑116). Bij de voltooiing van de studie was de mediane deelnameduur aan de studie inclusief safety follow‑upfase 60,9 maanden (spreiding: 0‑112,6). De totale incidentie van bevestigde ONJ gecorrigeerd voor patiëntjaren was 1,5 per 100 patiëntjaren (0,2 per 100 patiëntjaren tijdens het eerste jaar van de behandeling, 1,5 in het tweede jaar, 1,8 in het derde jaar, 2,1 in het vierde jaar, 1,4 in het vijfde jaar en daarna 2,2). De mediane tijd tot ONJ was 41 maanden (spreiding: 11‑96).
Studie 7 werd uitgevoerd om patiënten met GCTB die in studie 6 waren behandeld gedurende een extra periode van 5 jaar of langer te blijven volgen. ONJ werd gemeld bij 6 patiënten (11,8%) van de 51 blootgestelde patiënten met een mediaan totaal van 42 doses denosumab. Drie van deze gevallen van ONJ werden medisch bevestigd.
Geneesmiddelgerelateerde overgevoeligheidsreacties
In de postmarketingsetting zijn gevallen van overgevoeligheid, inclusief uitzonderlijke gevallen van anafylactische reacties, gerapporteerd bij patiënten die XGEVA toegediend kregen.
Atypische femurfracturen
In het klinisch onderzoeksprogramma in zijn geheel zijn atypische femurfracturen soms gemeld bij patiënten die behandeld werden met XGEVA en het risico nam toe naarmate de behandeling langer duurde. Deze fracturen traden op tijdens de behandeling en tot 9 maanden nadat de behandeling was stopgezet (zie rubriek 4.4).
In het klinisch onderzoeksprogramma voor GCTB zijn atypische femurfracturen vaak gemeld bij patiënten die behandeld werden met XGEVA. In studie 6 was de incidentie van bevestigde atypische femurfracturen 0,95% (5/526) bij patiënten met reusceltumor van het bot. In vervolgstudie 7 was de incidentie van bevestigde atypische femurfracturen 3,9% (2/51) van de aan denosumab blootgestelde patiënten.
Skeletspierstelselpijn
In de postmarketingsetting werd skeletspierstelselpijn, waaronder ernstige gevallen, gemeld bij patiënten die XGEVA kregen. In klinische onderzoeken kwam skeletspierstelselpijn zeer vaak voor in zowel de groep met denosumab als die met zoledroninezuur. Skeletspierstelselpijn die leidde tot stopzetting van de onderzoeksbehandeling kwam soms voor.
Nieuwe primaire maligniteit
In de primaire dubbelblinde behandelfases van vier actief gecontroleerde klinische fase III‑onderzoeken bij patiënten met gevorderde maligniteiten waarbij bot was betrokken, werd een nieuwe primaire maligniteit gerapporteerd bij 54 (1,5%) van de 3.691 met XGEVA behandelde patiënten (mediane blootstelling van 13,8 maanden; spreiding: 1,0‑51,7) en bij 33 (0,9%) van de 3.688 met zoledroninezuur behandelde patiënten (mediane blootstelling van 12,9 maanden; spreiding: 1,0‑50,8).
De cumulatieve incidentie na één jaar was respectievelijk 1,1% voor denosumab en 0,6% voor zoledroninezuur.
Er bestond geen duidelijk behandelingsgerelateerd patroon voor afzonderlijke vormen van kanker of kankergroepen.
Bij patiënten met reusceltumor van het bot was in studie 6 de incidentie van nieuwe maligniteit, met inbegrip van maligniteiten van het bot en buiten het bot, 3,8% (20/526). In vervolgstudie 7 was de incidentie 11,8% (6/51) van de aan denosumab blootgestelde patiënten.
Lichenoïde reacties door medicijngebruik
Lichenoïde reacties door medicijngebruik (bijvoorbeeld reacties die lijken op lichen planus) zijn gemeld bij patiënten in de postmarketingsetting.
Pediatrische patiënten
XGEVA werd beoordeeld in een open‑label onderzoek waarin 28 adolescenten met een volgroeid skelet en met reusceltumor van het bot waren opgenomen. Op basis van deze beperkte gegevens leek het bijwerkingenprofiel overeen te komen met dat van volwassenen.
Klinisch significante hypercalciëmie na het stopzetten van de behandeling is gemeld in de postmarketingsetting bij pediatrische patiënten (zie rubriek 4.4).
Andere speciale patiëntengroepen
Nierfunctiestoornis
In een klinisch onderzoek hadden patiënten zonder een gevorderde maligniteit met een ernstige nierfunctiestoornis (creatinineklaring < 30 ml/min) of patiënten met nierdialyse een verhoogd risico op het ontwikkelen van hypocalciëmie wanneer zij geen calciumsuppletie kregen. Het risico op de ontwikkeling van hypocalciëmie tijdens de behandeling met XGEVA stijgt naarmate de ernst van de nierinsufficiëntie toeneemt. In een klinisch onderzoek bij patiënten zonder gevorderde kanker trad, ondanks calciumsuppletie, hypocalciëmie op bij 19% van de patiënten met een ernstige nierfunctiestoornis (creatinineklaring < 30 ml/min) en bij 63% van de patiënten die met nierdialyse werden behandeld. De totale incidentie van klinisch significante hypocalciëmie was 9%.
Hierbij optredende verhogingen van de waarden voor parathyroïdhormoon werden eveneens waargenomen bij met XGEVA behandelde patiënten met ernstige nierinsufficiëntie of patiënten die met nierdialyse werden behandeld. De controle van calciumspiegels en voldoende inname van calcium en vitamine D is vooral belangrijk bij patiënten met een nierfunctiestoornis (zie rubriek 4.4).
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg.be
Luxemburg
Centre Régional de Pharmacovigilance de Nancy of Division de la pharmacie et des médicaments de la Direction de la santé
Website: www.guichet.lu/pharmacovigilance
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Amgen Europe B.V.
Minervum 7061,
4817 ZK Breda,
Nederland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/11/703/001
EU/1/11/703/002
EU/1/11/703/003
EU/1/11/703/004
EU/1/11/703/005
EU/1/11/703/006
10. DATUM VAN HERZIENING VAN DE TEKST
juli 2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 2883296 | XGEVA 120 MG OPLOSSING VOOR INJECTIE 1 FL | M05BX04 | € 161,13 | - | Ja | € 12,8 | € 8,5 |
| 2883304 | XGEVA 120 MG OPLOSSING VOOR INJECTIE 4 FL | M05BX04 | € 611,62 | - | Ja | € 12,8 | € 8,5 |
| 4792420 | XGEVA 120MG VOORGEVULDE SPUIT 4 | € 611,62 | - | Ja | € 12,8 | € 8,5 | |
| 4792438 | XGEVA 120MG VOORGEVULDE SPUIT 1 | € 161,13 | - | Ja | € 12,8 | € 8,5 |