SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Zejula 100 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke filmomhulde tablet bevat niraparibtosylaatmonohydraat, overeenkomend met 100 mg niraparib.
Hulpstoffen met bekend effect
Elke filmomhulde tablet bevat 34,7 mg lactosemonohydraat (zie rubriek 4.4).
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet).
Grijze, ovale (12 mm x 8 mm) filmomhulde tablet met aan de ene zijde de markering “100” en aan de andere zijde “Zejula”.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Zejula is geïndiceerd:
- als monotherapie voor de onderhoudsbehandeling van volwassen patiënten met gevorderde epitheliale hooggradige eierstok‑, eileider‑ of primaire peritoneumkanker (van FIGO‑stadia III en IV), die (volledig of partieel) reageren na afronding van een eerstelijnsbehandeling met op platina gebaseerde chemotherapie;
- als monotherapie voor de onderhoudsbehandeling van volwassen patiënten met platinagevoelige, gerecidiveerde, hooggradige sereuze epitheliale eierstok‑, eileider‑ of primaire peritoneumkanker, die (volledig of partieel) reageren op op platina gebaseerde chemotherapie.
4.2 Dosering en wijze van toediening
Behandeling met Zejula dient te worden gestart door en onder toezicht te staan van een arts die ervaring heeft met het gebruik van geneesmiddelen tegen kanker.
Dosering
Eerstelijnsonderhoudsbehandeling van eierstokkanker
De aanbevolen aanvangsdosis Zejula is 200 mg (twee tabletten van 100 mg) eenmaal daags. Voor patiënten die ≥ 77 kg wegen en bij baseline een bloedplaatjestelling ≥ 150.000/μl hebben, is de aanbevolen aanvangsdosis Zejula echter 300 mg (drie tabletten van 100 mg) eenmaal daags (zie rubrieken 4.4 en 4.8).
Onderhoudsbehandeling van gerecidiveerde eierstokkanker
De dosis is drie tabletten van 100 mg eenmaal daags, overeenkomend met een totale dagdosis van 300 mg.
Patiënten dienen te worden aangespoord om hun dosis elke dag ongeveer op hetzelfde tijdstip in te nemen. Toediening bij het naar bed gaan kan een potentiële methode zijn om misselijkheid te beheersen.
Het wordt aanbevolen de behandeling voort te zetten tot ziekteprogressie of toxiciteit optreedt.
Overgeslagen dosis
Als patiënten een dosis niet hebben ingenomen, dienen zij hun volgende dosis op het normale, geplande tijdstip in te nemen.
Dosisaanpassingen vanwege bijwerkingen
De aanbevolen dosisaanpassingen vanwege bijwerkingen staan vermeld in tabel 1, 2 en 3.
Over het algemeen wordt aanbevolen om de behandeling eerst te onderbreken (maar niet langer dan 28 dagen achter elkaar) om de patiënt te laten herstellen van de bijwerking, en vervolgens weer met dezelfde dosis te starten. Wanneer de bijwerking opnieuw optreedt, wordt aanbevolen de behandeling te onderbreken en vervolgens te hervatten met de verlaagde dosis. Als de bijwerking na een dosisonderbreking van 28 dagen nog steeds aanhoudt, wordt aanbevolen om te stoppen met Zejula. Als bijwerkingen niet kunnen worden behandeld met deze strategie van dosisonderbreking en ‑verlaging, wordt aanbevolen om te stoppen met Zejula.
Tabel 1: Aanbevolen dosisaanpassingen vanwege bijwerkingen
Niveau aanvangsdosis | 200 mg | 300 mg |
Eerste dosisverlaging | 100 mg/dag | 200 mg/dag (twee tabletten van 100 mg) |
Tweede dosisverlaging | Zejula stoppen. | 100 mg/daga (één tablet van 100 mg) |
a Stop met Zejula als de dosis verder moet worden verlaagd naar minder dan 100 mg/dag.
Tabel 2: Dosisaanpassingen vanwege niet hematologische bijwerkingen
Niet‑hematologische, behandelingsgerelateerde bijwerking van CTCAE‑graad ≥ 3, waarbij profylaxe niet haalbaar wordt geacht of de bijwerking ondanks behandeling blijft bestaan | Eerste optreden: |
Tweede optreden: | |
Behandelingsgerelateerde bijwerking van CTCAE‑graad ≥ 3 die langer dan 28 dagen aanhoudt terwijl de patiënt Zejula 100 mg/dag krijgt toegediend | Behandeling stoppen. |
CTCAE=Common Terminology Criteria for Adverse Events.
Tabel 3: Dosisaanpassingen vanwege hematologische bijwerkingen
Hematologische bijwerkingen zijn waargenomen tijdens behandeling met Zejula, vooral in de beginfase van de behandeling. Het wordt daarom aanbevolen om gedurende de eerste maand van de behandeling het volledige bloedbeeld wekelijks te controleren en de dosis indien nodig aan te passen. Na de eerste maand wordt aanbevolen om het volledige bloedbeeld maandelijks en daarna periodiek te controleren (zie rubriek 4.4). Gebaseerd op individuele laboratoriumwaarden kunnen wekelijkse controles tijdens de tweede maand gerechtvaardigd zijn. | |
Hematologische bijwerkingen waarvoor ondersteuning met transfusie of hematopoëtische groeifactor nodig is | • Voor patiënten met een aantal bloedplaatjes ≤ 10.000/μl dient een bloedplaatjestransfusie te worden overwogen. In geval van andere risicofactoren voor bloeding, zoals gelijktijdige toediening van anticoagulantia of trombocytenaggregatieremmers, dient onderbreking van de toediening van deze stoffen en/of transfusie met meer trombocyten te worden overwogen. |
Aantal bloedplaatjes < 100.000/μl | Eerste optreden: |
Tweede optreden: | |
Neutrofielen < 1.000/µl of hemoglobine < 5,0 mmol/l (8 g/dl) | • Zejula gedurende maximaal 28 dagen stoppen en wekelijks het volledige bloedbeeld controleren totdat het aantal neutrofielen is teruggekeerd tot ≥ 1.500/µl of de hemoglobinewaarde is teruggekeerd tot ≥ 5,6 mmol/l (9 g/dl). |
Bevestigde diagnose van myelodysplastisch syndroom (MDS) of acute myeloïde leukemie | • Permanent stoppen met Zejula. |
Patiënten met een laag lichaamsgewicht bij de onderhoudsbehandeling van gerecidiveerde eierstokkanker
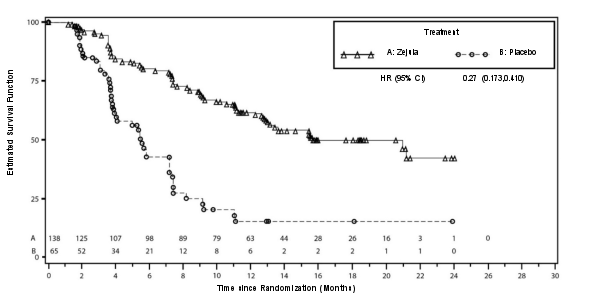
Ongeveer 25% van de patiënten in het NOVA‑onderzoek woog minder dan 58 kg, en ongeveer 25% van de patiënten woog meer dan 77 kg. De incidentie van bijwerkingen van graad 3 of 4 was onder patiënten met een laag lichaamsgewicht (78%) hoger dan onder patiënten met een hoog lichaamsgewicht (53%). Slechts 13% van de patiënten met een laag lichaamsgewicht bleef na cyclus 3 op een dosis van 300 mg. Een aanvangsdosis van 200 mg kan worden overwogen voor patiënten die minder dan 58 kg wegen.
Ouderen
Er is geen dosisaanpassing nodig voor oudere patiënten (≥ 65 jaar). Er zijn beperkte klinische gegevens over patiënten van 75 jaar en ouder.
Nierfunctiestoornis
Er is geen dosisaanpassing nodig voor patiënten met een lichte tot matige nierfunctiestoornis. Er zijn geen gegevens over patiënten met een ernstige nierfunctiestoornis of terminale nierziekte die hemodialyse ondergaan; bij deze patiënten dient voorzichtigheid te worden betracht (zie rubriek 5.2).
Leverfunctiestoornis
Er is geen dosisaanpassing nodig voor patiënten met een lichte leverfunctiestoornis (ofwel aspartaataminotransferase (ASAT) > bovengrens van de normaalwaarde (ULN) en totaal bilirubine (TB) ≤ ULN of elke ASAT en TB > 1,0 x ‑ 1,5 x ULN). Voor patiënten met een matige leverfunctiestoornis (elke ASAT en TB > 1,5 x ‑ 3 x ULN) is de aanbevolen startdosering van Zejula 200 mg eenmaal daags. Er zijn geen gegevens over patiënten met een ernstige leverfunctiestoornis (elke ASAT en TB > 3 x ULN); bij deze patiënten dient voorzichtigheid te worden betracht (zie rubrieken 4.4 en 5.2).
Patiënten met een Eastern Cooperative Oncology Group (ECOG)-performance status 2 tot 4
Er zijn geen klinische gegevens beschikbaar over patiënten met een ECOG‑prestatiestatus 2 tot 4.
Pediatrische patiënten
De veiligheid en werkzaamheid van Zejula bij kinderen en adolescenten met een leeftijd
jonger dan 18 jaar zijn niet vastgesteld. Buiten de goedgekeurde indicaties is Zejula in combinatie met dostarlimab onderzocht bij kinderen in de leeftijd van 5 tot minder dan 18 jaar met gerecidiveerde of refractaire solide tumoren waaronder osteosarcoom en neuroblastoom; de onderzoeksgegevens waren echter beperkt en op basis van de resultaten van het onderzoek kon niet geconcludeerd worden dat de voordelen van dergelijk gebruik zwaarder wegen dan de risico’s. De momenteel beschikbare gegevens worden beschreven in rubriek 5.1 en 5.2.
Wijze van toediening
Zejula is voor oraal gebruik.
Het wordt aanbevolen Zejula tabletten zonder voedsel in te nemen (ten minste 1 uur voor of 2 uur na een maaltijd) of met een lichte maaltijd (zie rubriek 5.2).
4.3 Contra‑indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
Borstvoeding (zie rubriek 4.6).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Bijwerkingen van alle graden die optraden bij ≥ 10% van de 851 patiënten die Zejula als monotherapie kregen in de gepoolde PRIMA- (aanvangsdosis van 200 mg of 300 mg) en NOVA‑onderzoeken: nausea, anemie, trombocytopenie, vermoeidheid, constipatie, braken, hoofdpijn, insomnia, plaatjestelling verlaagd, neutropenie, abdominale pijn, verminderde eetlust, diarree, dyspneu, hypertensie, asthenie, duizeligheid, neutrofielentelling verlaagd, hoesten, artralgie, rugpijn, wittebloedceltelling verlaagd en opvliegers.
De vaakst voorkomende ernstige bijwerkingen > 1% (frequenties van tijdens de behandeling opgetreden bijwerkingen) waren trombocytopenie en anemie.
Lijst van bijwerkingen in tabelvorm
De volgende bijwerkingen zijn vastgesteld op basis van klinische onderzoeken en postmarketing surveillance bij patiënten die Zejula als monotherapie kregen (zie tabel 4). De frequenties waarmee bijwerkingen optraden zijn gebaseerd op gepoolde gegevens van bijwerkingen die zijn gegenereerd uit de PRIMA- en NOVA-onderzoeken (vaste aanvangsdosis van 300 mg/dag) waarbij de blootstelling van de patiënt bekend is en als volgt gedefinieerd:
Zeer vaak: ≥ 1/10
Vaak: ≥ 1/100, < 1/10
Soms: ≥ 1/1.000, < 1/100
Zelden: ≥ 1/10.000, < 1/1.000
Zeer zelden: < 1/10.000
Binnen elke frequentiecategorie worden de bijwerkingen weergegeven in volgorde van afnemende ernst.
Tabel 4: Lijst van bijwerkingen in tabelvorm
Systeem/orgaanklasse | Frequentie van alle CTCAE‑graden | Frequentie van CTCAE‑graad 3 of 4 |
Infecties en parasitaire aandoeningen | Zeer vaak | Soms |
Neoplasmata, benigne, maligne en niet‑gespecificeerd (inclusief cysten en poliepen) | Vaak | Vaak |
Bloed‑ en lymfestelselaandoeningen | Zeer vaak | Zeer vaak |
Immuunsysteemaandoeningen | Vaak | Soms |
Voedings‑ en stofwisselingsstoornissen | Zeer vaak | Vaak |
Psychische stoornissen | Zeer vaak | Soms |
Zenuwstelselaandoeningen | Zeer vaak | Soms |
Hartaandoeningen | Zeer vaak |
|
Bloedvataandoeningen | Zeer vaak | Vaak |
Ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen | Zeer vaak | Soms |
Maagdarmstelselaandoeningen | Zeer vaak | Vaak |
Huid‑ en onderhuidaandoeningen | Vaak | Soms |
Skeletspierstelsel‑ en bindweefselaandoeningen | Zeer vaak | Soms |
Algemene aandoeningen en toedieningsplaatsstoornissen | Zeer vaak | Vaak |
Onderzoeken | Vaak | Vaak |
CTCAE = Common Terminology Criteria for Adverse Events versie 4.02.
a Op basis van gegevens uit klinisch onderzoek naar niraparib. Deze zijn niet beperkt tot het hoofdonderzoek ENGOT-OV16 met monotherapie.
b Omvat overgevoeligheid, overgevoeligheid voor geneesmiddelen, anafylactoïde reactie, geneesmiddeleruptie, angio-oedeem en urticaria.
c Omvat geheugenstoornis, concentratiestoornis.
De bijwerkingen die werden gezien bij de groep patiënten die een aanvangsdosis Zejula van 200 mg kregen toegediend op basis van gewicht of aantal bloedplaatjes bij baseline, kwamen even vaak of minder vaak voor in vergelijking met de groep die een vaste aanvangsdosis van 300 mg kreeg toegediend (tabel 4).
Zie hieronder voor specifieke informatie over de frequentie van trombocytopenie, anemie en neutropenie.
Beschrijving van geselecteerde bijwerkingen
Hematologische bijwerkingen (trombocytopenie, anemie, neutropenie), waaronder klinische diagnoses en/of laboratoriumuitslagen, traden doorgaans vroeg in de behandeling met niraparib op, waarbij de incidentie in de loop van de tijd afnam.
In NOVA en PRIMA hadden patiënten die in aanmerking kwamen voor behandeling met Zejula de volgende uitgangswaarden van hematologische parameters: absoluut aantal neutrofielen (ANC) ≥ 1.500 cellen/µl; bloedplaatjes ≥ 100.000 cellen/µl en hemoglobine ≥ 5,6 mmol/l (9 g/dl; NOVA) of ≥ 6,2 mmol/l (10 g/dl;PRIMA) vóór behandeling. In het klinische programma werden hematologische bijwerkingen behandeld met bewaking van de laboratoriumwaarden en dosisaanpassingen (zie rubriek 4.2).
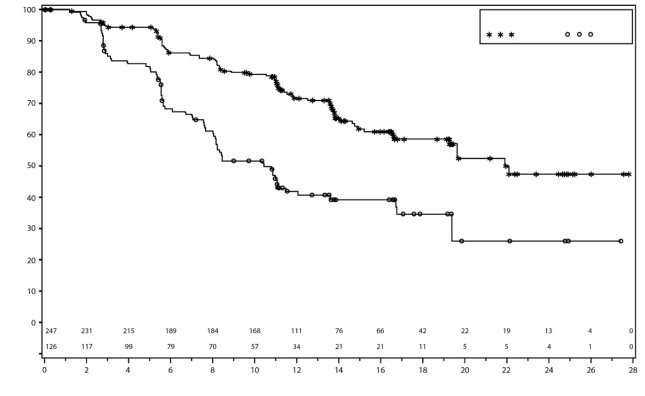
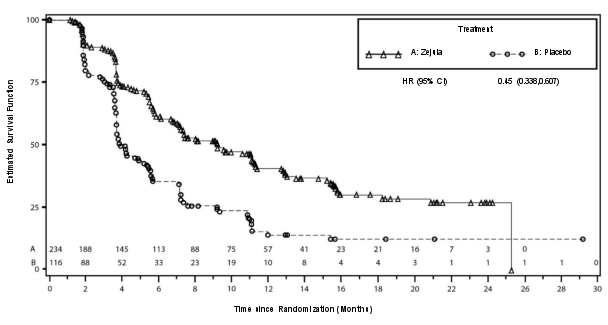
In PRIMA, bij patiënten die een aanvangsdosis Zejula kregen toegediend op basis van gewicht of aantal bloedplaatjes bij baseline, waren trombocytopenie, anemie en neutropenie van graad ≥ 3 verminderd van respectievelijk 48% naar 21%, 36% naar 23% en 24% naar 15%, vergeleken met de groep die een vaste aanvangsdosis van 300 mg kreeg toegediend. De frequentie hiervan was lager in vergelijking met de groep die een vaste aanvangsdosis van 300 mg kreeg toegediend. Stoppen vanwege trombocytopenie, anemie en neutropenie kwam voor bij respectievelijk 3%, 3% en 2% van de patiënten.
Trombocytopenie
In PRIMA kreeg ongeveer 39% van de patiënten die met Zejula werden behandeld trombocytopenie van graad 3/4 in vergelijking met 0,4% van de patiënten die met placebo werden behandeld, met een mediane tijd van eerste dosis tot eerste optreden van 22 dagen (spreiding: 15 tot 335 dagen) en met een mediane duur van 6 dagen (spreiding: 1 tot 374 dagen). Stoppen vanwege trombocytopenie kwam voor bij 4% van de patiënten die niraparib kregen.
In NOVA kreeg ongeveer 60% van de patiënten trombocytopenie van enige graad, en 34% van de patiënten kreeg trombocytopenie graad 3/4. Bij patiënten met minder dan 180 × 109/l trombocyten bij baseline kwamen trombocytopenie van enige graad en trombocytopenie graad 3/4 voor bij respectievelijk 76% en 45% van de patiënten. De mediane tijd tot het ontstaan van trombocytopenie ongeacht de graad en trombocytopenie graad 3/4 was respectievelijk 22 en 23 dagen. Het percentage nieuwe gevallen van trombocytopenie na intensieve dosisaanpassingen die werden uitgevoerd gedurende de eerste twee maanden van behandeling vanaf cyclus 4 bedroeg 1,2%. De mediane duur van voorvallen van trombocytopenie ongeacht de graad was 23 dagen, en de mediane duur van trombocytopenie graad 3/4 was 10 dagen. Patiënten die worden behandeld met Zejula en trombocytopenie ontwikkelen, kunnen een verhoogd risico van hemorragie hebben. In het klinische programma werd trombocytopenie behandeld met bewaking van laboratoriumwaarden, dosisaanpassing en bloedplaatjestransfusie indien aangewezen (zie rubriek 4.2). Stoppen vanwege voorvallen van trombocytopenie (trombocytopenie en aantal bloedplaatjes verlaagd) kwam voor bij ongeveer 3% van de patiënten.
In NOVA hadden 13% (48/367) van de patiënten bloeding met gelijktijdige trombocytopenie; alle bloedingsvoorvallen die gelijktijdig met trombocytopenie optraden, waren van graad 1 of 2 in ernst, met uitzondering van één voorval van petechiën en hematoom van graad 3 dat gelijktijdig met een ernstige bijwerking van pancytopenie werd waargenomen. Trombocytopenie kwam vaker voor bij patiënten met een aantal bloedplaatjes minder dan 180 × 109/l bij baseline. Ongeveer 76% van de patiënten met lagere aantallen bloedplaatjes bij baseline (< 180 × 109/l) die Zejula ontvingen, kreeg trombocytopenie ongeacht de graad en 45% van de patiënten kreeg trombocytopenie van graad 3/4. Pancytopenie is waargenomen bij < 1% van de patiënten die niraparib kregen.
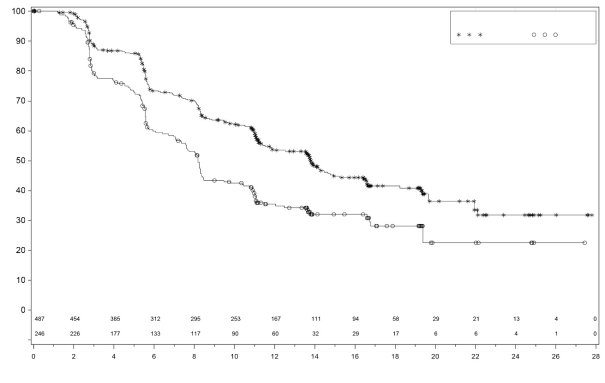
Anemie
In PRIMA kreeg 31% van de patiënten die met Zejula werden behandeld anemie van graad 3/4 in vergelijking met 2% van de patiënten die placebo kregen, met een mediane tijd van eerste dosis tot eerste optreden van 80 dagen (spreiding: 15 tot 533 dagen) en met een mediane duur van 7 dagen (spreiding: 1 tot 119 dagen). Stoppen vanwege anemie kwam voor bij 2% van de patiënten die niraparib kregen.
In NOVA kreeg ongeveer 50% van de patiënten anemie van enige graad, en 25% kreeg anemie graad 3/4. De mediane tijd tot het ontstaan van anemie ongeacht de graad was 42 dagen, en 85 dagen voor voorvallen van graad 3/4. De mediane duur van anemie ongeacht de graad was 63 dagen, en 8 dagen voor voorvallen van graad 3/4. Anemie van elke graad kan tijdens de behandeling met Zejula aanhouden. In het klinische programma werd anemie behandeld met bewaking van de laboratoriumwaarden, dosisaanpassing (zie rubriek 4.2) en indien aangewezen met transfusies met rode bloedcellen. Stoppen vanwege anemie kwam voor bij 1% van de patiënten.
Neutropenie
In PRIMA kreeg 21% van de patiënten die met Zejula werden behandeld neutropenie van graad 3/4 in vergelijking met 1% van de patiënten die placebo kregen, met een mediane tijd van eerste dosis tot eerste optreden van 29 dagen (spreiding: 15 tot 421 dagen) en met een mediane duur van 8 dagen (spreiding: 1 tot 42 dagen). Stoppen vanwege neutropenie kwam voor bij 2% van de patiënten die niraparib kregen.
In NOVA kreeg ongeveer 30% van de patiënten neutropenie van enige graad, en 20% van de patiënten kreeg neutropenie graad 3/4. De mediane tijd tot het ontstaan van neutropenie ongeacht de graad was 27 dagen, en 29 dagen voor voorvallen van graad 3/4. De mediane duur van neutropenie ongeacht de graad was 26 dagen, en 13 dagen voor voorvallen van graad 3/4. Daarnaast werd granulocyt‑koloniestimulerende factor (GCSF) toegediend aan ongeveer 6% van de patiënten die met niraparib werden behandeld, als gelijktijdige behandeling van neutropenie. Stoppen vanwege voorvallen van neutropenie kwam voor bij 2% van de patiënten.
Myelodysplastisch syndroom/acute myeloïde leukemie
Tijdens klinische onderzoeken trad MDS/AML op bij 1% van de patiënten die werden behandeld met Zejula. Van die gevallen had 41% een dodelijke afloop. Na 75 maanden follow‑up voor overleving was de incidentie hoger bij patiënten met recidiverende eierstokkanker die 2 of meer lijnen eerdere chemotherapie met platina hadden gekregen en die gBRCAmut hadden. Alle patiënten hadden mogelijk bijdragende factoren voor de ontwikkeling van MDS/AML, aangezien ze eerdere chemotherapie hadden gekregen met platinamiddelen. Velen hadden ook andere DNA‑beschadigende middelen en radiotherapie ontvangen. Het merendeel van de meldingen kwam van gBRCAmut‑dragers. Sommige patiënten hadden een voorgeschiedenis van eerdere kanker of beenmergsuppressie.
In PRIMA was de incidentie van MDS/AML 2,3% bij patiënten die Zejula kregen en 1,6% bij patiënten die placebo kregen met een follow-up van 74 maanden.
In NOVA bij patiënten met recidiverende eierstokkanker die ten minste twee eerdere lijnen chemotherapie met platina hadden gekregen, was de algehele incidentie van MDS/AML 3,8% bij patiënten die Zejula kregen en 1,7% bij patiënten die placebo kregen met een follow‑up van 75 maanden. In gBRCAmut‑ en non‑gBRCAmut‑cohorten was de incidentie van MDS/AML respectievelijk 7,4% en 1,7% bij patiënten die Zejula kregen en 3,1% en 0,9% bij patiënten die placebo kregen.
Hypertensie
In PRIMA kwam hypertensie van graad 3/4 voor bij 6% van de patiënten die met Zejula werden behandeld in vergelijking met 1% van de patiënten die placebo kregen, met een mediane tijd van eerste dosis tot eerste optreden van 50 dagen (spreiding: 1 tot 589 dagen) en met een mediane duur van 12 dagen (spreiding: 1 tot 61 dagen). Geen van de patiënten is gestopt met Zejula vanwege hypertensie.
In NOVA kwam hypertensie ongeacht de graad voor bij 19,3% van de patiënten die met Zejula werden behandeld. Hypertensie graad 3/4 kwam voor bij 8,2% van de patiënten. Hypertensie kon goed worden behandeld met antihypertensiva. Stoppen vanwege hypertensie kwam voor bij < 1% van de patiënten.
Melding van vermoedelijk bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
GlaxoSmithKline Trading Services Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Ierland
D24 YK11
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/17/1235/004
EU/1/17/1235/005
EU/1/17/1235/006
EU/1/17/1235/007
10. DATUM VAN HERZIENING VAN DE TEKST
04/06/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4763561 | ZEJULA 100MG FILMOMH TABL KINDVEILIGE BLISTER 56 | - | € 3800 | Ja | - | - |