SAMENVATTING VAN DE PRODUCTKENMERKEN
▼Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
1. NAAM VAN HET GENEESMIDDEL
Padcev 20 mg poeder voor concentraat voor oplossing voor infusie
Padcev 30 mg poeder voor concentraat voor oplossing voor infusie
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Padcev 20 mg poeder voor concentraat voor oplossing voor infusie
Eén injectieflacon met poeder voor concentraat voor oplossing voor infusie bevat 20 mg enfortumab vedotin.
Padcev 30 mg poeder voor concentraat voor oplossing voor infusie
Eén injectieflacon met poeder voor concentraat voor oplossing voor infusie bevat 30 mg enfortumab vedotin.
Na reconstitutie bevat elke ml oplossing 10 mg enfortumab vedotin.
Enfortumab vedotin bestaat uit een volledig humaan IgG1-kappa-antilichaam, geconjugeerd aan het microtubuli-ontregelende middel monomethylauristatine E (MMAE) door middel van een protease‑splitsbare maleimidocaproyl-valine‑citrulline-linker.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Poeder voor concentraat voor oplossing voor infusie.
Wit tot gebroken wit gevriesdroogd poeder.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Spierinvasief blaascarcinoom (MIBC)
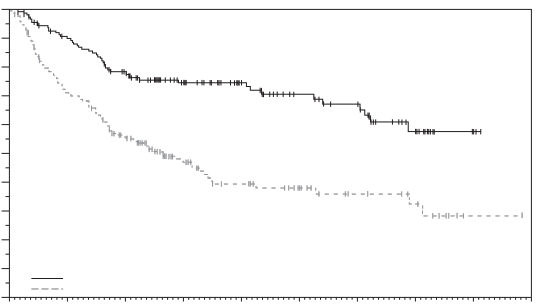
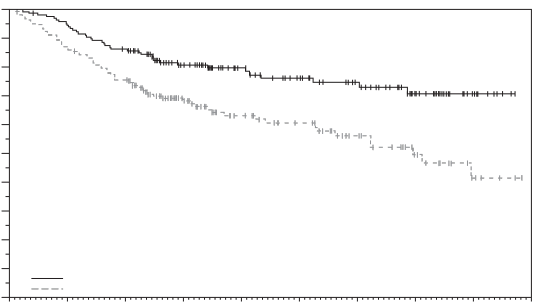
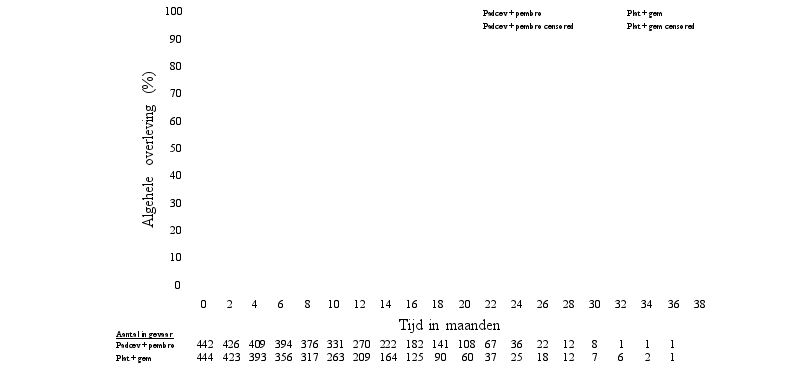
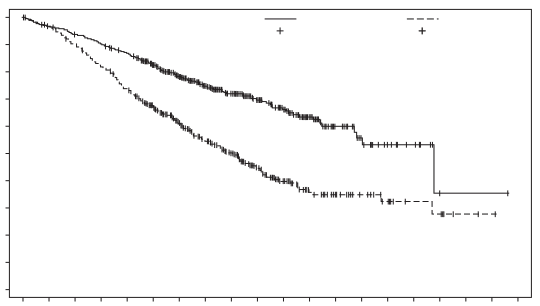
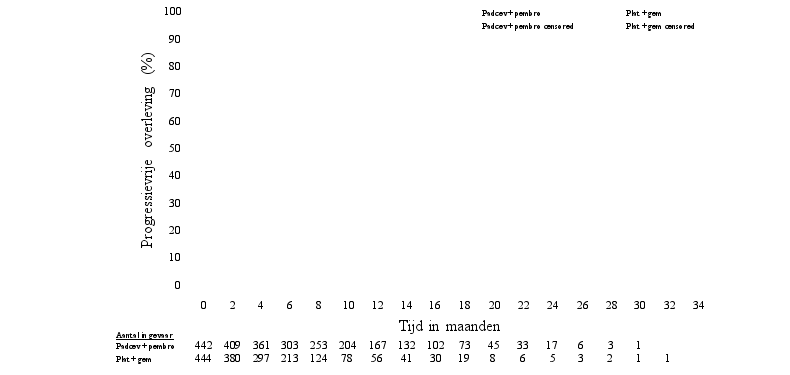
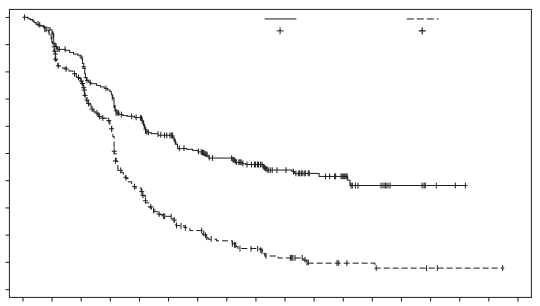
Padcev, in combinatie met pembrolizumab, als neoadjuvante behandeling en vervolgens voortgezet na radicale cystectomie als adjuvante behandeling, is geïndiceerd voor de behandeling van volwassen patiënten met reseceerbaar spierinvasief blaascarcinoom (muscle invasive bladder cancer, MIBC) die niet in aanmerking komen voor cisplatinebevattende chemotherapie.
Niet-reseceerbaar of gemetastaseerd urotheelcarcinoom
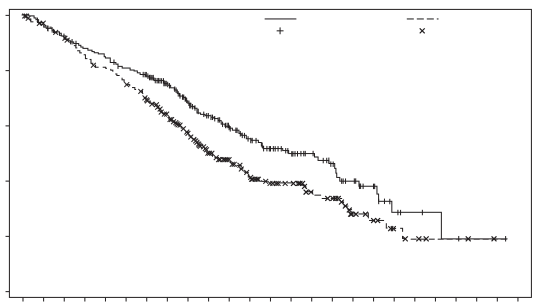
Padcev, in combinatie met pembrolizumab, is geïndiceerd voor de eerstelijnsbehandeling van volwassen patiënten met niet-reseceerbaar of gemetastaseerd urotheelcarcinoom die in aanmerking komen voor platinumbevattende chemotherapie.
Lokaal gevorderd of gemetastaseerd urotheelcarcinoom
Padcev als monotherapie is geïndiceerd voor de behandeling van volwassen patiënten met lokaal gevorderd of gemetastaseerd urotheelcarcinoom die eerder een platinumbevattende chemotherapie hebben ondergaan en een geprogrammeerde celdoodreceptor‑1- of geprogrammeerde celdoodligand 1-remmer hebben gekregen (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
De behandeling met Padcev dient te worden gestart onder toezicht van een arts die ervaring heeft met de toepassing van therapieën tegen kanker. Zorg voor goede veneuze toegang alvorens de behandeling te beginnen (zie rubriek 4.4).
Dosering
Tabel 1. Aanbevolen dosis voor enfortumab vedotin in combinatie met pembrolizumab*
Indicatie | Aanbevolen dosis enfortumab vedotin | Behandelingsduur |
Neoadjuvant en adjuvant spierinvasief blaascarcinoom (MIBC) | Enfortumab vedotin 1,25 mg/kg (tot max. 125 mg voor patiënten ≥ 100 kg) op dag 1 en 8 van een 21-daagse cyclus. | Patiënten die niet in aanmerking komen voor cisplatine |
Niet-reseceerbaar of gemetastaseerd urotheelcarcinoom | Enfortumab vedotin 1,25 mg/kg (tot max. 125 mg voor patiënten ≥ 100 kg) op dag 1 en 8 van een 21-daagse cyclus. | Totdat ziekteprogressie of onaanvaardbare toxiciteit optreedt. |
*Bij toediening op dezelfde dag dient de patiënt pembrolizumab na enfortumab vedotin te krijgen. Zie de Samenvatting van de productkenmerken van pembrolizumab voor aanvullende toedieningsinformatie van pembrolizumab.
Tabel 2. Aanbevolen dosis voor enfortumab vedotin als monotherapie
Indicatie | Aanbevolen dosis enfortumab vedotin | Behandelingsduur |
Lokaal gevorderd of gemetastaseerd urotheelcarcinoom | Enfortumab vedotin 1,25 mg/kg (tot max. 125 mg voor patiënten ≥ 100 kg) op dag 1, 8 en 15 van een 28-daagse cyclus. | Totdat ziekteprogressie of onaanvaardbare toxiciteit optreedt. |
Tabel 3. Aanbevolen dosisverlagingen van enfortumab vedotin bij bijwerkingen | |
Dosisverlagingsschema | Dosisniveau |
Aanvangsdosis | 1,25 mg/kg tot max. 125 mg |
Eerste dosisverlaging | 1,0 mg/kg tot max. 100 mg |
Tweede dosisverlaging | 0,75 mg/kg tot max. 75 mg |
Derde dosisverlaging | 0,5 mg/kg tot max. 50 mg |
Dosisaanpassingen
Tabel 4. Dosisonderbreking, -verlaging en -stopzetting van enfortumab vedotin | |||
Bijwerking | Ernst* | Dosisaanpassing* | |
Huidreacties | Vermoeden van Stevens‑Johnson-syndroom (SJS) of toxische epidermale necrolyse (TEN) of bulleuze laesies | Onderbreek onmiddellijk en verwijs door naar gespecialiseerde zorg. | |
Bevestigd(e) SJS of TEN; graad 4 of recidiverend graad 3 | Definitief stopzetten. | ||
Graad 2 verslechtering |
| ||
Hyperglykemie | Bloedglucose |
| |
Pneumonitis/ | Graad 2 |
| |
Graad ≥ 3 | Definitief stopzetten. | ||
Perifere neuropathie | Graad 2 |
| |
Graad ≥ 3 | Definitief stopzetten. | ||
*Toxiciteit is beoordeeld volgens de ‘Common Terminology Criteria’ voor bijwerkingen van het National Cancer Institute versie 5.0 (NCI-CTCAE v5.0), waarbij graad 1 licht is, graad 2 matig ernstig, graad 3 ernstig en graad 4 levensbedreigend. | |||
Speciale populaties
Ouderen
Er is geen dosisaanpassing nodig bij patiënten van ≥ 65 jaar (zie rubriek 5.2).
Nierfunctiestoornis
Er is geen dosisaanpassing nodig bij patiënten met een lichte (creatinineklaring [CrCL] > 60-90 ml/min), matig ernstige (CrCL 30-60 ml/min) of ernstige (CrCL 15-<30 ml/min) nierfunctiestoornis. Enfortumab vedotin is niet geëvalueerd bij patiënten met eindstadium nierziekte (CrCL < 15 ml/min) (zie rubriek 5.2).
Leverfunctiestoornis
Er is geen dosisaanpassing nodig bij patiënten met een lichte leverfunctiestoornis (totaal bilirubine 1 tot 1,5 × bovengrens van normaal [ULN] en elke ASAT, of totaal bilirubine ≤ ULN en ASAT > ULN). Enfortumab vedotin is geëvalueerd bij slechts een beperkt aantal patiënten met een matig ernstige of ernstige leverfunctiestoornis. Leverfunctiestoornissen zullen naar verwachting de systemische blootstelling aan MMAE (het cytotoxische geneesmiddel) verhogen; patiënten moeten daarom nauwlettend worden gecontroleerd op mogelijke bijwerkingen. Door beperkte data bij patiënten met matige en ernstige leverfunctiestoornissen kan er geen specifieke dosis worden aanbevolen (zie rubriek 5.2).
Pediatrische patiënten
Er is geen relevante toepassing van enfortumab vedotin bij pediatrische patiënten voor de indicatie lokaal gevorderd of gemetastaseerd urotheelcarcinoom en MIBC.
Wijze van toediening
Padcev is voor intraveneus gebruik. De aanbevolen dosering dient te worden toegediend via een intraveneus infuus gedurende 30 minuten. Enfortumab vedotin dient niet te worden toegediend als intraveneuze push- of bolusinjectie.
Voor instructies over reconstitutie en verdunning van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof(fen) of voor een van de in rubriek 6.1 vermelde hulpstof(fen).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Enfortumab vedotin in combinatie met pembrolizumab
Raadpleeg de samenvatting van de productkenmerken van pembrolizumab wanneer enfortumab vedotin wordt toegediend in combinatie met pembrolizumab voordat de behandeling wordt gestart.
Spierinvasief blaascarcinoom
De veiligheid van enfortumab vedotin werd beoordeeld in combinatie met pembrolizumab bij 167 patiënten die ten minste één dosis enfortumab vedotin 1,25 mg/kg kregen in combinatie met pembrolizumab in een fase 3-studie (EV-303) (zie tabel 5). Patiënten werden gedurende een mediane duur van 6,3 maanden blootgesteld aan enfortumab vedotin in combinatie met pembrolizumab (spreiding: 0,03 tot 19,7 maanden). De mediane duur van blootstelling aan alleen enfortumab vedotin was 5,5 maanden (spreiding: 0,03 tot 14,1 maanden).
De meest voorkomende bijwerkingen bij enfortumab vedotin in combinatie met pembrolizumab waren pruritus (47,3%), alopecia (34,7%), diarree (34,1%), vermoeidheid (32,3%), anemie (30,5%), verminderde eetlust (28,1%), dysgeusie (28,1%), nausea (25,7%), rash (25,1%), aspartaataminotransferase verhoogd (24%), gewicht verlaagd (19,8%), alanine-aminotransferase verhoogd (19,2%), rash maculopapulair (16,2%), droge huid (15%), hypothyreoïdie (14,4%), perifere sensorische neuropathie (13,8%), hyperglykemie (12,6%) en perifere neuropathie (10,2%).
De meest voorkomende ernstige bijwerking (≥ 2%) was diarree (2,4%). Eenenveertig procent van de patiënten stopte permanent met enfortumab vedotin vanwege bijwerkingen; de meest voorkomende bijwerking (≥ 2%) die leidde tot stopzetting, was perifere sensorische neuropathie (2,4%).
Bij 44% van de patiënten traden bijwerkingen op die leidden tot onderbreking van de behandeling met enfortumab vedotin. De meest voorkomende bijwerkingen (≥ 2%) die leidden tot onderbreking van de behandeling waren diarree (4,2%), rash (4,2%), neutropenie (3,6%), vermoeidheid (3%), hyperglykemie (3%) en jeuk (2,4%)
Bijwerkingen die leidden tot het verlagen van de dosis van enfortumab vedotin, kwamen voor bij 17% van de patiënten. De meest voorkomende bijwerkingen (≥ 2%) die leidden tot dosisverlaging, waren rash (2,4%) en gewicht verlaagd (2,4%).
Niet-reseceerbaar of gemetastaseerd carcinoom
De veiligheid van enfortumab vedotin werd beoordeeld in combinatie met pembrolizumab bij 564 patiënten die ten minste één dosis enfortumab vedotin 1,25 mg/kg kregen in combinatie met pembrolizumab in één fase 2-studie (EV-103) en één fase 3-studie (EV-302) (zie tabel 5). Patiënten werden gedurende een mediane duur van 9,4 maanden blootgesteld aan enfortumab vedotin in combinatie met pembrolizumab (bereik: 0,3 tot 34,4 maanden).
De meest voorkomende bijwerkingen bij enfortumab vedotin in combinatie met pembrolizumab waren perifere sensorische neuropathie (53,4%), pruritus (41,1%), vermoeidheid (40,4%), diarree (39,2%), alopecia (38,5%), rash maculopapulair (36%), gewicht verlaagd (36%), verminderde eetlust (33,9%), nausea (28,4%), anemie (25,7%), dysgeusie (24,3%), droge huid (18,1%), alanine-aminotransferase verhoogd (16,8%), hyperglykemie (16,7%), aspartaataminotransferase verhoogd (15,4%), droog oog (14,4%), braken (13,3%), rash maculair (11,3%), hypothyreoïdie (10,5%) en neutropenie (10,1%).
De meest voorkomende ernstige bijwerkingen (≥ 2%) waren diarree (3%) en pneumonitis (2,3%). Zesendertig procent van de patiënten stopte permanent met enfortumab vedotin vanwege bijwerkingen; de meest voorkomende bijwerkingen (≥ 2%) die leidden tot stopzetting, waren perifere sensorische neuropathie (12,2%) en rash maculopapulair (2%).
Bijwerkingen die leidden tot onderbreken van de toediening van enfortumab vedotin, kwamen voor bij 72% van de patiënten. De meest voorkomende bijwerkingen (≥ 2%) die leidden tot onderbreken van de toediening waren perifere sensorische neuropathie (17%), rash maculopapulair (6,9%), diarree (4,8%), vermoeidheid (3,7%), pneumonitis (3,7%), hyperglykemie (3,4%), neutropenie (3,2%), alanine-aminotransferase verhoogd (3%), pruritus (2,3%) en anemie (2%).
Bijwerkingen die leidden tot dosisverlaging van enfortumab vedotin, kwamen voor bij 42,4% van de patiënten. De meest voorkomende bijwerkingen (≥ 2%) die leidden tot dosisverlaging, waren perifere sensorische neuropathie (9,9%), rash maculopapulair (6,4%), vermoeidheid (3,2%), diarree (2,3%) en neutropenie (2,1%).
Enfortumab vedotin als monotherapie
De veiligheid van enfortumab vedotin werd beoordeeld als monotherapie bij 793 patiënten die ten minste één dosis enfortumab vedotin 1,25 mg/kg kregen in twee fase 1-onderzoeken (EV-101 en EV-102), drie fase 2-onderzoeken (EV-103, EV-201 en EV-203) en één fase 3-onderzoek (EV-301) (zie tabel 5). Patiënten werden gedurende een mediane duur van 4,7 maanden blootgesteld aan enfortumab vedotin (bereik: 0,3 tot 55,7 maanden).
De meest voorkomende bijwerkingen bij enfortumab vedotin waren alopecia (47,7%), verminderde eetlust (47,2%), vermoeidheid (46,8%), diarree (39,1%), perifere sensorische neuropathie (38,5%), nausea (37,8%), pruritus (33,4%), dysgeusie (30,4%), anemie (29,1%), gewicht verlaagd (25,2%), rash maculopapulair (23,6%), droge huid (21,8%), braken (18,7%), aspartaataminotransferase verhoogd (17%), hyperglykemie (14,9%), droog oog (12,7%), alanine-aminotransferase verhoogd (12,7%) en rash (11,6%).
De meest voorkomende ernstige bijwerkingen (≥ 2%) waren diarree (2,1%) en hyperglykemie (2,1%). Eenentwintig procent van de patiënten stopte permanent met enfortumab vedotin vanwege bijwerkingen; de meest voorkomende bijwerking (≥ 2%) die leidde tot stopzetting, was perifere sensorische neuropathie (4,8%). Bijwerkingen die leidden tot onderbreken van de dosering, kwamen voor bij bij 62% van de patiënten; de meest voorkomende bijwerkingen (≥ 2%) die leidden tot onderbreken van de dosering waren perifere sensorische neuropathie (14,8%), vermoeidheid (7,4%), rash maculopapulair (4%), aspartaataminotransferase verhoogd (3,4%), alanineaminotransferase verhoogd (3,2%), anemie (3,2%), hyperglykemie (3,2%), neutrofielentelling verlaagd (3%), diarree (2,8%), rash (2,4%) en perifere motorische neuropathie (2,1%). Achtendertig procent van de patiënten had een dosisverlaging nodig vanwege een bijwerking; de meest voorkomende bijwerkingen (≥ 2%) die leidden tot een dosisverlaging waren perifere sensorische neuropathie (10,3%), vermoeidheid (5,3%), rash maculopapulair (4,2%) en verminderde eetlust (2,1%).
Lijst van bijwerkingen in tabelvorm
Bijwerkingen die zijn waargenomen tijdens klinische onderzoeken met enfortumab vedotin als monotherapie of in combinatie met pembrolizumab, of gemeld tijdens postmarketing gebruik van enfortumab vedotin, worden in deze rubriek weergegeven per frequentiecategorie. De frequentiecategorieën zijn als volgt gedefinieerd: zeer vaak (≥1/10); vaak (≥1/100, <1/10); soms (≥1/1 000, <1/100); zelden (≥1/10 000, <1/1 000); zeer zelden (<1/10 000); niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiegroep worden de bijwerkingen gepresenteerd in volgorde van afnemende ernst.
Tabel 5. Bijwerkingen bij patiënten behandeld met enfortumab vedotin | ||
Frequentie | Monotherapie | In combinatie met pembrolizumab |
Infecties en parasitaire aandoeningen | ||
Vaak | Sepsis, pneumonie | Sepsis, pneumonie |
Bloed- en lymfestelselaandoeningen | ||
Zeer vaak | Anemie | Anemie |
Vaak | Trombocytopenie | Trombocytopenie |
Niet bekend1 | Neutropenie, febriele neutropenie, neutrofielentelling verlaagd | Neutropenie, febriele neutropenie, neutrofielentelling verlaagd |
Endocriene aandoeningen | ||
Zeer vaak |
| Hypothyroïdie |
Voedings- en stofwisselingsstoornissen | ||
Zeer vaak | Hyperglykemie, verminderde eetlust | Hyperglykemie, verminderde eetlust |
Niet bekend1 | Diabetische ketoacidose | Diabetische ketoacidose |
Zenuwstelselaandoeningen | ||
Zeer vaak | Perifere sensorische neuropathie, dysgeusie | Perifere sensorische neuropathie, dysgeusie |
Vaak | Perifere neuropathie, perifere motorische neuropathie, perifere sensomotorische neuropathie, paresthesie, hypo-esthesie, loopstoornis, spierzwakte | Perifere neuropathie, perifere motorische neuropathie, perifere sensomotorische neuropathie, paresthesie, hypo-esthesie, loopstoornis, spierzwakte, neurotoxiciteit |
Soms | Demyeliniserende polyneuropathie, polyneuropathie, neurotoxiciteit, motorische disfunctie, dysesthesie, spieratrofie, neuralgie, peroneale zenuwverlamming, gevoelsverlies, brandend gevoel van de huid, branderig gevoel | Dysesthesie, polyneuropathie, myasthenia gravis, neuralgie, peroneale zenuwverlamming, brandend gevoel van de huid |
Oogaandoeningen | ||
Zeer vaak | Droog oog | Droog oog |
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen | ||
Vaak | Pneumonitis/ILD2 | Pneumonitis/ILD2 |
Maagdarmstelselaandoeningen | ||
Zeer vaak | Diarree, braken, nausea | Diarree, braken, nausea |
Huid- en onderhuidaandoeningen | ||
Zeer vaak | Alopecia, pruritus, rash, rash maculopapulair, droge huid | Alopecia, pruritus, rash maculopapulair, droge huid |
Vaak | Geneesmiddeleneruptie, huidexfoliatie, conjunctivitis, bulleuze dermatitis, blaar, stomatitis, palmoplantair erytrodysesthesiesyndroom, eczeem, erytheem, rash erythemateus, rash maculair, rash papulair, rash pruritisch, rash vesiculair | Rash, huidexfoliatie, conjunctivitis, bulleuze dermatitis, blaar, stomatitis, palmoplantair erytrodysesthesie-syndroom, eczeem, erytheem, rash erythemateus, rash maculair, rash papulair, rash pruritisch, rash vesiculair, dermatitis |
Soms | Gegeneraliseerde exfoliatieve dermatitis, erythema multiforme, exfoliatieve rash, pemfigoïd, rash maculovesiculair, dermatitis, dermatitis allergisch, contactdermatitis, intertrigo, huidirritatie, stasisdermatitis, bloedblaar | Geneesmiddeleneruptie, gegeneraliseerde exfoliatieve dermatitis, erythema multiforme, exfoliatieve rash, pemfigoïd, dermatitis allergisch, contactdermatitis, huidirritatie, stasisdermatitis |
Niet bekend1 | Toxische epidermale necrolyse, hyperpigmentatie van de huid, huidverkleuring, pigmentatiestoornis, syndroom van Stevens-Johnson, epidermale necrose, symmetrisch geneesmiddelgerelateerd intertrigineus en flexuraal exantheem | Toxische epidermale necrolyse, hyperpigmentatie van de huid, huidverkleuring, pigmentatiestoornis, syndroom van Stevens-Johnson, epidermale necrose, symmetrisch geneesmiddelgerelateerd intertrigineus en flexuraal exantheem |
Skeletspierstelsel- en bindweefselaandoeningen | ||
Soms |
| Myositis |
Algemene aandoeningen en toedieningsplaatsstoornissen | ||
Zeer vaak | Vermoeidheid | Vermoeidheid |
Vaak | Extravasatie op infuusplaats | Extravasatie op infuusplaats |
Onderzoeken | ||
Zeer vaak | Alanineaminotransferase verhoogd, aspartaataminotransferase verhoogd, gewicht verlaagd | Alanineaminotransferase verhoogd, aspartaataminotransferase verhoogd, gewicht verlaagd |
Vaak |
| Lipase verhoogd |
Letsel, intoxicaties en verrichtingencomplicaties | ||
Vaak | Infusiegerelateerde reactie | Infusiegerelateerde reactie |
1 Gebaseerd op postmarketingervaring wereldwijd. | ||
Beschrijving van geselecteerde bijwerkingen
Immunogeniciteit
In totaal werden 159 patiënten onderzocht op immunogeniciteit voor enfortumab vedotin na enfortumab vedotin in combinatie met pembrolizumab voor de behandeling van MIBC; bij 3 patiënten werd bevestigd dat ze bij baseline positief waren voor antidrugantilichamen (ADA), en van de patiënten die bij baseline negatief waren (n=156), waren er na baseline in totaal 2 (1,3%) positief.
In totaal 490 patiënten werden onderzocht op immunogeniciteit voor enfortumab vedotin na enfortumab vedotin in combinatie met pembrolizumab voor de behandeling van niet-reseceerbaar of gemetastaseerd urotheelcarcinoom; bij 24 patiënten werd bevestigd dat ze bij baseline positief waren voor ADA, en van de patiënten die bij baseline negatief waren (n=466), waren er na baseline in totaal 14 (3%) positief.
In totaal 697 patiënten werden onderzocht op immunogeniciteit voor 1,25 mg/kg enfortumab vedotin als monotherapie; bij 16 patiënten werd bevestigd dat ze bij baseline positief waren voor antidrugantilichamen (ADA), en van de patiënten die bij baseline negatief waren (n=681), waren er na baseline in totaal 24 (3,5%) positief.
De incidentie van tijdens de behandeling optredende anti-enfortumab vedotin-antilichaamvorming was consistent bij beoordeling na toediening van enfortumab vedotin als monotherapie en in combinatie met pembrolizumab.
Vanwege het beperkte aantal patiënten met antilichamen tegen Padcev kunnen er geen conclusies worden getrokken over een eventueel effect van immunogeniciteit op werkzaamheid, veiligheid of farmacokinetiek.
Huidreacties
In klinische onderzoeken naar enfortumab vedotin in combinatie met pembrolizumab kwamen huidreacties voor bij 89% (648) van de 731 patiënten en een meerderheid van deze huidreacties omvatte rash maculo-papulair, rash maculair, rash en rash papulair. Ernstige (graad 3 of 4) huidreacties kwamen voor bij 19% (135) van de patiënten (graad 3: 17%, graad 4: 2%). Bij één patiënt deed zich een fataal voorval van toxische epidermale necrolyse voor. De mediane tijd tot de aanvang van ernstige huidreacties was 1,6 maanden (bereik: 0,1 tot 17,2 maanden). Van de patiënten bij wie huidreacties voorkwamen en van wie er gegevens waren over resolutie (n = 496), had 76% volledige resolutie, 16% gedeeltelijke verbetering en 8% geen verbetering ten tijde van hun laatste beoordeling. Van de 24% van de patiënten met residuele huidreacties bij de laatste beoordeling had 25% bijwerkingen van graad ≥ 2.
In klinische onderzoeken met enfortumab vedotin als monotherapie, kwamen huidreacties voor bij 57% (452) van de 793 patiënten die werden behandeld met 1,25 mg/kg enfortumab vedotin. Ernstige (graad 3 of 4) huidreacties kwamen voor bij 14% (108) van de patiënten en het merendeel van deze reacties omvatte rash maculopapulair, stomatitis, erythemateuze huiduitslag, huiduitslag of geneesmiddeleneruptie. De mediane tijd tot de aanvang van ernstige huidreacties was 0,7 maanden (bereik: 0,1 tot 8,2 maanden). Ernstige huidreacties kwamen voor bij 4,3% (34) van de patiënten. Van de patiënten bij wie huidreacties voorkwamen en van wie er gegevens waren over resolutie (n=366), had 61% volledige resolutie, 24% gedeeltelijke verbetering en 15% geen verbetering ten tijde van hun laatste beoordeling. Van de 39% van de patiënten met residuele huidreacties bij de laatste beoordeling had 38% bijwerkingen van graad ≥ 2.
Pneumonitis/ILD
In klinische onderzoeken met enfortumab vedotin in combinatie met pembrolizumab kwam pneumonitis/ILD voor bij 9,4% (69) van de 731 patiënten. Ernstige (graad 3 of 4) pneumonitis/ILD kwam voor bij 20 patiënten (graad 3: 2,3%, graad 4: 0,4%). Pneumonitis/ILD leidde tot stopzetting van enfortumab vedotin bij 2,1% van de patiënten. Bij drie patiënten deed zich een fataal voorval voor van pneumonitis/ILD. De mediane tijd tot de aanvang van pneumonitis/ILD, ongeacht graad, was 4,1 maanden (bereik: 0,3 tot 26,2 maanden).
In klinische onderzoeken met enfortumab vedotin als monotherapie kwam pneumonitis/ILD voor bij 3,3% (26) van de 793 patiënten die werden behandeld met 1,25 mg/kg enfortumab vedotin. Minder dan 1% van de patiënten kreeg ernstige (graad 3 of 4) pneumonitis/ILD (graad 3: 0,5%, graad 4: 0,3%). Pneumonitis/ILD leidde tot stopzetting van enfortumab vedotin bij 0,5% van de patiënten. Er waren geen sterfgevallen ten gevolge van pneumonitis/ILD. De mediane tijd tot het optreden van pneumonitis/ ILD, ongeacht graad, was 2,7 maanden (bereik: 0,6 tot 6,0 maanden) en de mediane duur was 1,6 maanden (bereik: 0,1 tot 43,0 maanden). Van de 26 patiënten die pneumonitis/ILD kregen, verdwenen bij 8 (30,8%) de symptomen volledig.
Hyperglykemie
In klinische onderzoeken met enfortumab vedotin als monotherapie kwam hyperglykemie (bloedglucose > 13,9 mmol/l) voor bij 17% (133) van de 793 patiënten die werden behandeld met 1,25 mg/kg enfortumab vedotin. Ernstige gevallen van hyperglykemie kwamen voor bij 2,5% van de patiënten, 7% van de patiënten kreeg ernstige (graad 3 of 4) hyperglykemie en bij 0,3% van de patiënten deed zich een fataal voorval voor; één betrof hyperglykemie en één betrof diabetische ketoacidose. De incidentie van hyperglykemie graad 3‑4 nam overeenkomstig toe bij patiënten met een hogere BMI en bij patiënten met een hogere waarde voor hemoglobine A1C (HbA1c) bij baseline. De mediane tijd tot de aanvang van hyperglykemie was 0,5 maanden (bereik: 0 tot 20,3 maanden). Van de patiënten bij wie hyperglykemie voorkwam en van wie er gegevens waren over resolutie ervan (n=106), had 66% volledige resolutie, 19% gedeeltelijke verbetering en 15% geen verbetering ten tijde van hun laatste beoordeling. Van de 34% patiënten met residuele hyperglykemie bij de laatste beoordeling had 64% bijwerkingen van graad ≥ 2.
Perifere neuropathie
In klinische onderzoeken met enfortumab vedotin als monotherapie deed zich perifere neuropathie voor bij 53% (422) van de 793 patiënten die werden behandeld met enfortumab vedotin 1,25 mg/kg. Vijf procent van de patiënten kreeg ernstige (graad 3 of 4) perifere neuropathie, waaronder sensorische en motorische voorvallen. De mediane tijd tot de aanvang van graad ≥ 2 perifere neuropathie was 5 maanden (bereik: 0,1 tot 20,2 maanden). Van de patiënten bij wie neuropathie voorkwam en van wie er gegevens waren over resolutie ervan (n=340), had 14% volledige resolutie, 46% gedeeltelijke verbetering en 41% geen verbetering ten tijde van hun laatste beoordeling. Van de 86% van de patiënten met residuele neuropathie bij de laatste beoordeling had 51% bijwerkingen van graad ≥ 2.
Oculaire aandoeningen
In klinische onderzoeken met enfortumab vedotin als monotherapie ervoer 30% van de patiënten droge ogen tijdens de behandeling met 1,25 mg/kg enfortumab vedotin. De behandeling werd onderbroken bij 1,5% van de patiënten en 0,1% van de patiënten stopte definitief met de behandeling vanwege droge ogen. Ernstige (graad 3) droge ogen deden zich voor bij slechts 3 patiënten (0,4%). De mediane tijd tot de aanvang van droge ogen was 1,7 maanden (bereik: 0 tot 30,6 maanden).
Speciale patiëntengroepen
Ouderen
Enfortumab vedotin in combinatie met pembrolizumab is onderzocht bij 202 patiënten < 65 jaar en 529 patiënten ≥ 65 jaar. Over het algemeen was de frequentie van bijwerkingen hoger bij patiënten ≥ 65 jaar vergeleken met patiënten < 65 jaar, met name van ernstige bijwerkingen (respectievelijk 58,8% en 40,1%) en bijwerkingen van ≥ graad 3 (respectievelijk 79,8% en 65,3%).
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be



7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Astellas Pharma Europe B.V.
Sylviusweg 62
2333 BE Leiden
Nederland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/21/1615/001
EU/1/21/1615/002
10. DATUM VAN HERZIENING VAN DE TEKST
19/06/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 4566196 | PADCEV 20MG PDR OPL INF FL 1 | - | € 600 | Ja | - | - | |
| 4566204 | PADCEV 30MG PDR OPL INF FL 1 | - | € 900 | Ja | - | - |