1. NAAM VAN HET GENEESMIDDEL
Xtandi 40 mg filmomhulde tabletten
Xtandi 80 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Xtandi 40 mg filmomhulde tabletten
Elke filmomhulde tablet bevat 40 mg enzalutamide.
Xtandi 80 mg filmomhulde tabletten
Elke filmomhulde tablet bevat 80 mg enzalutamide.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet.
Xtandi 40 mg filmomhulde tabletten
Gele ronde - filmomhulde tabletten, met de inscriptie E 40.
Xtandi 80 mg filmomhulde tabletten
Gele ovale - filmomhulde tabletten, met de inscriptie E 80.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Xtandi is geïndiceerd:
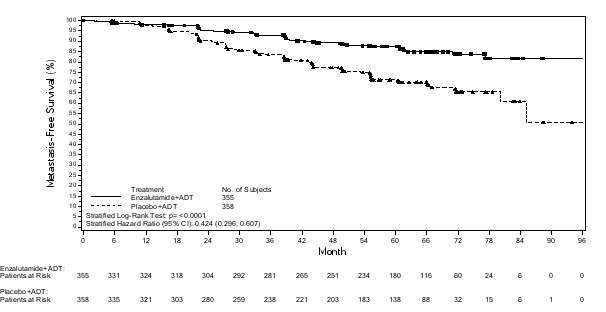
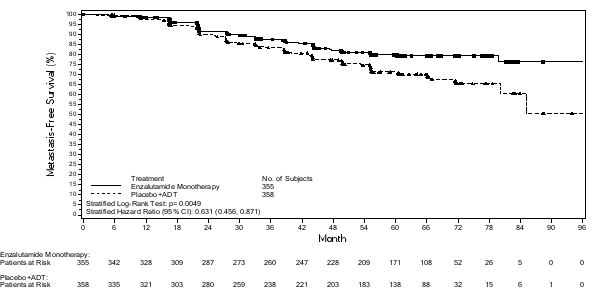
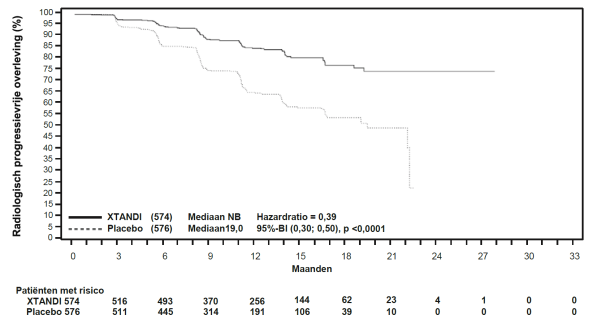
- als monotherapie of in combinatie met androgeendeprivatietherapie voor de behandeling van volwassen mannen met niet-gemetastaseerd, hormoongevoelig prostaatcarcinoom (nmHSPC) met een hoog risico biochemisch recidief (BCR) die niet in aanmerking komen voor salvage-radiotherapie (zie rubriek 5.1).
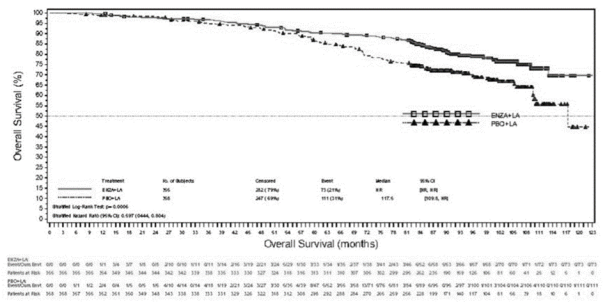
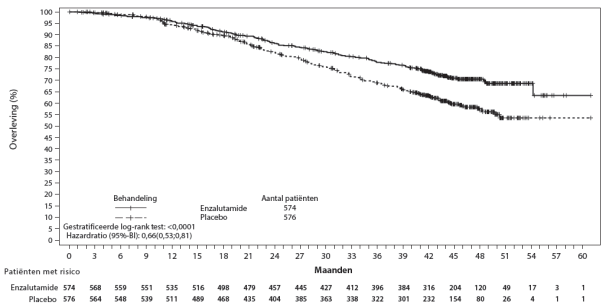
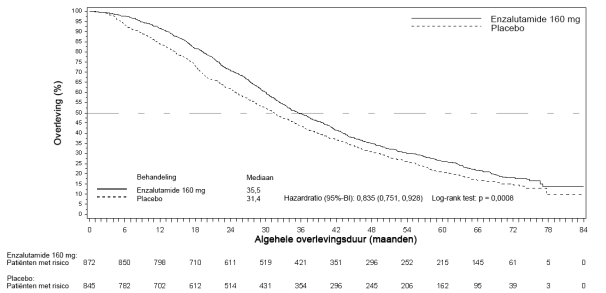
- in combinatie met androgeendeprivatietherapie voor de behandeling van volwassen mannen met gemetastaseerd, hormoongevoelig prostaatcarcinoom (mHSPC) (zie rubriek 5.1).
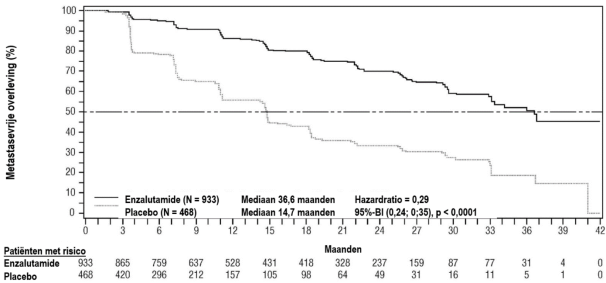
- voor de behandeling van volwassen mannen met niet-gemetastaseerd hoogrisico-CRPC (castratieresistent prostaatcarcinoom) (zie rubriek 5.1).
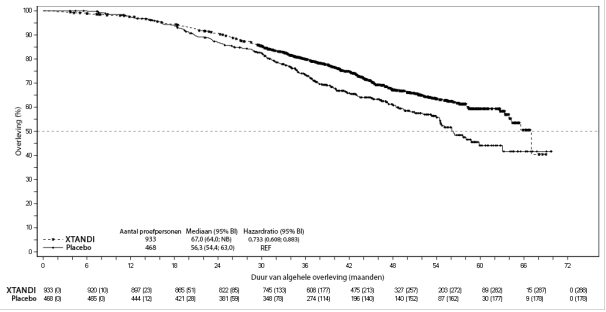
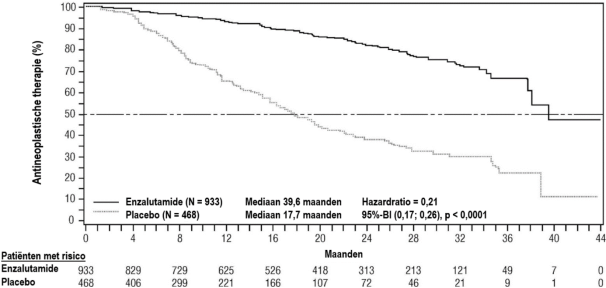
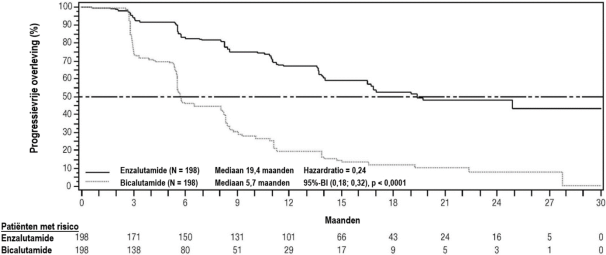
- voor de behandeling van volwassen mannen met gemetastaseerd CRPC die asymptomatisch of licht symptomatisch zijn na falen van androgeendeprivatietherapie voor wie behandeling met chemotherapie nog niet klinisch geïndiceerd is (zie rubriek 5.1).
- voor de behandeling van volwassen mannen met gemetastaseerd CRPC bij wie de ziekte progressief was tijdens of na behandeling met docetaxel.
4.2 Dosering en wijze van toediening
Behandeling met enzalutamide moet worden gestart en gemonitord door gespecialiseerde artsen die ervaring hebben met de medische behandeling van prostaatkanker.
Dosering
De aanbevolen dosis is 160 mg enzalutamide (vier filmomhulde tabletten van 40 mg of twee filmomhulde tabletten van 80 mg) als eenmaal daagse orale dosis.
Bij patiënten met CRPC of mHSPC die niet operatief zijn gecastreerd dient chemische castratie met een ‘Luteinising Hormone-Releasing Hormone’ (LHRH)-analoog tijdens de behandeling te worden voortgezet.
Patiënten met nmHSPC met een hoog risico BCR kunnen worden behandeld met Xtandi met of zonder een LHRH-analoog. Bij patiënten die Xtandi ontvangen met of zonder een LHRH-analoog, kan de behandeling worden onderbroken indien de PSA niet-detecteerbaar (< 0,2 ng/ml) is na 36 weken therapie. De behandeling dient te worden hervat wanneer de PSA is toegenomen tot ≥ 2,0 ng/ml bij patiënten die eerder een radicale prostatectomie hebben ondergaan, of ≥ 5,0 ng/ml bij patiënten die eerder primaire radiotherapie hebben ondergaan. Indien de PSA na 36 weken therapie detecteerbaar is (≥ 0,2 ng/ml) dient de behandeling te worden voortgezet (zie rubriek 5.1).
Als een patiënt Xtandi niet op het gebruikelijke tijdstip inneemt, dient de voorgeschreven dosis zo dicht mogelijk op het gebruikelijke tijdstip te worden ingenomen. Als een patiënt een dosis van een hele dag mist, dient de behandeling de volgende dag met de gebruikelijke dagelijkse dosis te worden hervat.
Als een patiënt last krijgt van een ≥ graad 3 toxiciteit of een onverdraaglijke bijwerking, dient de behandeling gestopt te worden gedurende één week of tot de symptomen verbeteren tot ≤ graad 2. Vervolgens dient de behandeling, indien gerechtvaardigd, hervat te worden op dezelfde of een verlaagde dosis (120 mg of 80 mg).
Gelijktijdig gebruik met sterke CYP2C8-remmers
Het gelijktijdig gebruik van sterke CYP2C8-remmers dient, indien mogelijk, te worden vermeden. Als aan patiënten ook een sterke CYP2C8-remmer dient te worden toegediend, dient de dosis enzalutamide verlaagd te worden naar 80 mg eenmaal daags. Als het gelijktijdig toedienen van de sterke CYP2C8-remmer wordt stopgezet, dient de dosis enzalutamide weer teruggebracht te worden naar de dosis zoals deze was voorafgaand aan het toedienen van de sterke CYP2C8-remmer (zie rubriek 4.5).
Ouderen
Er is geen dosisaanpassing noodzakelijk voor oudere patiënten (zie rubrieken 5.1 en 5.2).
Leverinsufficiëntie
Er is geen dosisaanpassing noodzakelijk voor patiënten met lichte, matige of ernstige leverinsufficiëntie (respectievelijk Child-Pugh-klasse A, B of C). Een toegenomen halfwaardetijd van enzalutamide is echter waargenomen bij patiënten met ernstige leverinsufficiëntie (zie rubrieken 4.4 en 5.2).
Nierinsufficiëntie
Er is geen dosisaanpassing noodzakelijk voor patiënten met lichte of matige nierinsufficiëntie (zie rubriek 5.2). Voorzichtigheid is geboden bij patiënten met ernstige nierinsufficiëntie of terminale nierziekte (zie rubriek 4.4).
Pediatrische patiënten
Er is geen relevante toepassing van enzalutamide bij pediatrische patiënten voor de indicatie behandeling van volwassen mannen met CRPC, mHSPC of nmHSPC met een hoog risico BCR.
Wijze van toediening
Xtandi is voor oraal gebruik. De filmomhulde tabletten Xtandi mogen niet worden versneden, verpulverd of gekauwd, maar moeten in hun geheel worden doorgeslikt met voldoende water en kunnen met of zonder voedsel worden ingenomen.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof(fen) of voor een van de in rubriek 6.1 vermelde hulpstof(fen).
Vrouwen die zwanger zijn of kunnen worden (zie rubrieken 4.6 en 6.6).
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
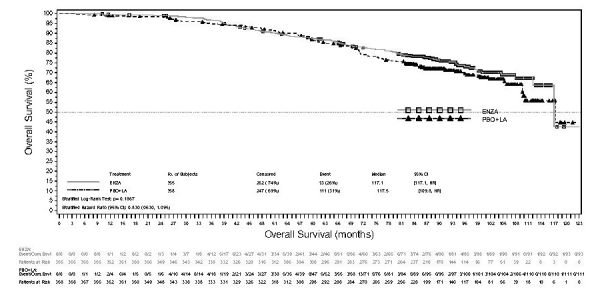
De meest voorkomende bijwerkingen zijn asthenie/vermoeidheid, opvliegers, hypertensie, fracturen, vallen en hoofdpijn. Tot andere belangrijke bijwerkingen behoren ischemische hartziekte en insulten.
Insulten traden op bij 0,6% van de met enzalutamide behandelde patiënten, bij 0,1% van de met placebo behandelde patiënten en bij 0,3% van de met bicalutamide behandelde patiënten.
Zeldzame gevallen van het posterieure reversibele encefalopathiesyndroom zijn gerapporteerd bij patiënten die zijn behandeld met enzalutamide (zie rubriek 4.4).
Het Stevens-Johnson-syndroom is gemeld bij behandeling met enzalutamide (zie rubriek 4.4).
Lijst met bijwerkingen in tabelvorm
De bijwerkingen waargenomen tijdens klinische studies worden hieronder per frequentiecategorie opgesomd. De frequentiecategorieën van bijwerkingen worden als volgt gedefinieerd: zeer vaak (≥1/10), vaak (≥1/100, <1/10), soms (≥1/1.000, <1/100), zelden (≥1/10.000, <1/1.000), zeer zelden (<1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Binnen elke frequentiegroep zijn de bijwerkingen gerangschikt op afnemende ernst.
Tabel 1: Bijwerkingen die zijn vastgesteld in de gecontroleerde klinische studies en post-marketing
MedDRA Systeem/orgaanklasse | Bijwerking en frequentie |
Bloed- en lymfestelselaandoeningen | Soms: leukopenie, neutropenie |
Immuunsysteemaandoeningen | Niet bekend*: gelaatsoedeem, tongoedeem, lipoedeem, farynxoedeem |
Voedings- en stofwisselingsstoornissen | Niet bekend*: verminderde eetlust |
Psychische stoornissen | Vaak: angst |
Zenuwstelselaandoeningen | Zeer vaak: hoofdpijn |
Hartaandoeningen | Vaak: ischemische hartziekte† |
Bloedvataandoeningen | Zeer vaak: opvlieger, hypertensie |
Maagdarmstelselaandoeningen | Niet bekend*: dysfagie∞, misselijkheid, braken, diarree |
Lever- en galaandoeningen | Soms: verhoogde leverenzymwaarden |
Huid- en onderhuidaandoeningen | Vaak: droge huid, pruritus |
Skeletspierstelsel- en bindweefselaandoeningen | Zeer vaak: fracturen‡ |
Voortplantingsstelsel- en borstaandoeningen | Vaak: gynaecomastie, tepelpijn#, borstgevoeligheid# |
Algemene aandoeningen en toedieningsplaatsstoornissen | Zeer vaak: asthenie, vermoeidheid |
Letsels, intoxicaties en verrichtingscomplicaties | Zeer vaak: vallen |
* Spontane meldingen afkomstig van post-marketingervaring.
¥ Geëvalueerd aan de hand van nauw begrensde zoekbewerkingen (narrow SMQ) van ‘Convulsies’ met inbegrip van convulsie, ‘grand mal’-convulsie, complexe partiële insulten, partiële insulten en status epilepticus. Dit omvat ook zeldzaam voorkomende insulten met complicaties die overlijden tot gevolg hebben.
† Geëvalueerd aan de hand van nauw begrensde zoekbewerkingen (narrow SMQ) van ‘Myocardinfarct’ en ‘Andere ischemische hartziekte’ met inbegrip van de volgende voorkeurstermen waargenomen bij ten minste twee patiënten in gerandomiseerde, placebogecontroleerde fase 3-onderzoeken: angina pectoris, kransslagaderaandoening, myocardinfarcten, acuut myocardinfarct, acuut coronairsyndroom, instabiele angina pectoris, myocardischemie en kransslagaderateriosclerose.
‡ Omvat alle voorkeurstermen met het woord ‘fractuur’ bij botten.
# Bijwerkingen bij enzalutamide als monotherapie
∞ Er zijn meldingen geweest van dysfagie, waaronder meldingen van verstikking. Beide voorvallen werden voornamelijk gemeld bij het innemen van het geneesmiddel in capsulevorm, wat verband zou kunnen houden met een grotere productgrootte (zie rubriek 4.4).
Beschrijving van geselecteerde bijwerkingen
Insult
In gecontroleerde klinische studies kregen 31 patiënten (0,6%) van de 5.112 patiënten die behandeld werden met een dagelijkse dosis van 160 mg enzalutamide een insult, terwijl vier patiënten (0,1%) die behandeld werden met placebo en één patiënt (0,3%) die behandeld werd met bicalutamide een insult kregen. De dosis lijkt een belangrijke voorspeller van het risico op insult te zijn, zoals weergegeven in preklinische gegevens en gegevens uit een dosisescalatiestudie. In de gecontroleerde klinische studies werden patiënten met een eerder insult of risicofactoren voor het krijgen van een insult uitgesloten.
In de single-armstudie 9785-CL-0403 (UPWARD) om de incidentie van insulten te beoordelen bij patiënten met predisponerende factoren voor een insult (waarbij 1,6% een voorgeschiedenis van insulten had), kregen 8 (2,2%) van de 366 patiënten die met enzalutamide behandeld werden, een insult. De mediane duur van de behandeling was 9,3 maanden.
Het mechanisme waardoor enzalutamide de insultdrempel kan verlagen is niet bekend, maar kan te maken hebben met gegevens uit in-vitro-onderzoeken waaruit blijkt dat enzalutamide en de actieve metaboliet ervan zich binden aan en de activiteit kunnen remmen van het GABA-gereguleerde chloridekanaal.
Ischemische hartziekte
In gerandomiseerde, placebogecontroleerde klinische studies trad ischemische hartziekte op bij 3,5% van de patiënten die werden behandeld met enzalutamide plus androgeendeprivatietherapie (ADT) vergeleken met 2,1% van de patiënten die werden behandeld met placebo plus ADT. Vijftien (0,4%) patiënten die werden behandeld met enzalutamide plus ADT en 3 (0,1%) patiënten die werden behandeld met placebo plus ADT kregen een ischemische hartziektevoorval dat overlijden tot gevolg had.
In de EMBARK-studie trad ischemische hartziekte op bij 6,2% van de patiënten die werden behandeld met enzalutamide plus leuproreline, en bij 10,7% van de patiënten die werden behandeld met enzalutamide als monotherapie. Eén (0,3%) van de patiënten die werden behandeld met enzalutamide plus leuproreline, één (0,3%) van de patiënten die werden behandeld met placebo plus leuproreline en één (0,3%) van de patiënten die werden behandeld met enzalutamide als monotherapie had een ischemisch hartziektevoorval met overlijden tot gevolg.
Gynaecomastie
In de EMBARK-studie werd gynaecomastie (alle graden) gezien bij 31 van de 353 patiënten (8,8%) die werden behandeld met enzalutamide plus leuproreline, en bij 163 van de 354 patiënten (46%) die werden behandeld met enzalutamide als monotherapie. Er werd geen gynaecomastie van graad 3 of hoger gezien bij patiënten die werden behandeld met enzalutamide plus leuproreline; dit werd wel gezien bij 1 van de patiënten (0,3%) die werden behandeld met placebo plus leuproreline en 3 van de patiënten (0,8%) die werden behandeld met enzalutamide als monotherapie.
Tepelpijn
In de EMBARK-studie werd tepelpijn (alle graden) gezien bij 13 van de 353 patiënten (3,7%) die werden behandeld met enzalutamide plus leuproreline, en bij 54 van de 354 patiënten (15,3%) die werden behandeld met enzalutamide als monotherapie. Er werd geen tepelpijn van graad 3 of hoger gezien bij patiënten die werden behandeld met enzalutamide plus leuproreline of met enzalutamide als monotherapie.
Borstgevoeligheid
In de EMBARK-studie werd borstgevoeligheid (alle graden) gezien bij 4 van de 353 patiënten (1,1%) die werden behandeld met enzalutamide plus leuproreline, en bij 51 van de 354 patiënten (14,4%) die werden behandeld met enzalutamide als monotherapie. Er werd geen borstgevoeligheid van graad 3 of hoger gezien bij patiënten die werden behandeld met enzalutamide plus leuproreline of met enzalutamide als monotherapie.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem.
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Astellas Pharma Europe B.V.
Sylviusweg 62
2333 BE Leiden
Nederland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/13/846/002 (filmomhulde tablet 40 mg)
EU/1/13/846/003 (filmomhulde tablet 80 mg)
10. DATUM VAN HERZIENING VAN DE TEKST
12/02/2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3899929 | XTANDI 40 MG FILMOMH TABL 112 X 40MG | L02BB04 | - | € 2800,84 | Ja | - | - |