SAMENVATTING VAN DE PRODUCTKENMERKEN
![]() Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg worden verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
Dit geneesmiddel is onderworpen aan aanvullende monitoring. Daardoor kan snel nieuwe veiligheidsinformatie worden vastgesteld. Beroepsbeoefenaren in de gezondheidszorg worden verzocht alle vermoedelijke bijwerkingen te melden. Zie rubriek 4.8 voor het rapporteren van bijwerkingen.
1. NAAM VAN HET GENEESMIDDEL
Benlysta 200 mg oplossing voor injectie in voorgevulde pen.
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Iedere voorgevulde pen van 1 ml bevat 200 mg belimumab.
Belimumab is een humaan, IgG1λ-monoklonaal antilichaam, geproduceerd door middel van recombinant-DNA-techniek in een cellijn (NS0), afkomstig van zoogdieren.
Hulpstof met bekend effect
Iedere voorgevulde pen bevat 0,1 mg polysorbaat 80.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Oplossing voor injectie in voorgevulde pen (injectie).
Een heldere tot opaalachtige, kleurloze tot lichtgele oplossing met een pH van 6 en een osmolaliteit van 270 - 320 mOsm/kg.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Benlysta is geïndiceerd als toegevoegde therapie bij patiënten van 5 jaar en ouder met actieve, auto-antilichaampositieve systemische lupus erythematosus (SLE) met een hoge mate van ziekteactiviteit (bijvoorbeeld positief anti-dsDNA en laag complement), ondanks een standaardbehandeling (zie rubriek 5.1).
Benlysta is geïndiceerd in combinatie met immunosuppressieve achtergrondbehandelingen voor de behandeling van volwassen patiënten met actieve lupusnefritis (zie rubriek 4.2 en 5.1).
4.2 Dosering en wijze van toediening
Behandeling met Benlysta mag alleen gestart en onder toezicht begeleid worden door een gekwalificeerde arts die ervaring heeft in de diagnosestelling en de behandeling van SLE.
Er wordt aanbevolen om de eerste subcutane injectie met Benlysta toe te dienen onder supervisie van een gekwalificeerde medische beroepsbeoefenaar en in een omgeving waar directe hulp kan worden geboden in geval van overgevoeligheidsreacties. De medische beroepsbeoefenaar moet de juiste training geven in de subcutane techniek en voldoende uitleg geven over klachten en symptomen van overgevoeligheidsreacties (zie rubriek 4.4). Een patiënt kan zelf injecteren of de verzorger van de patiënt kan Benlysta toedienen nadat de medische beroepsbeoefenaar heeft vastgesteld dat dit op de juiste manier gebeurt.
Bij patiënten jonger dan 10 jaar moet de Benlysta voorgevulde pen worden toegediend door een medische beroepsbeoefenaar of een daartoe getrainde verzorger.
Dosering
SLE
De toestand van de patiënt moet voortdurend beoordeeld worden. Als er na 6 maanden behandeling geen verbetering in de controle van de aandoening is opgetreden, moet staken van de behandeling met Benlysta overwogen worden.
Volwassenen
De aanbevolen dosering is 200 mg Benlysta eenmaal per week, subcutaan toegediend. De dosering is niet gebaseerd op gewicht (zie rubriek 5.2).
Kinderen en adolescenten (in de leeftijd van 5 tot 18 jaar)
De aanbevolen subcutane dosis is gebaseerd op het lichaamsgewicht (zie rubriek 5.1 en 5.2).
Lichaamsgewicht | Aanbevolen dosis |
≥ 50 kg | 200 mg eenmaal per week |
30 tot < 50 kg | 200 mg elke 10 dagen |
15 tot < 30 kg | 200 mg elke 2 weken |
Lupusnefritis
Volwassenen
Bij patiënten die starten met een therapie met Benlysta voor actieve lupusnefritis, is het aanbevolen doseringsschema eenmaal per week een dosis van 400 mg (twee injecties van 200 mg) gedurende 4 doses, dan eenmaal per week 200 mg daarna. Bij patiënten die doorgaan met therapie met Benlysta voor actieve lupusnefritis, is de aanbevolen dosering eenmaal per week 200 mg. Benlysta moet worden gebruikt in combinatie met corticosteroïden en mycofenolaat of cyclofosfamide voor inductie of met mycofenolaat of azathioprine voor onderhoud. De toestand van de patiënt moet voortdurend beoordeeld worden.
Gemiste doses
Indien een dosis wordt gemist, wordt aanbevolen om deze zo snel mogelijk toe te dienen. Daarna kunnen patiënten de toediening hervatten op de gebruikelijke dag van toediening, of met een nieuw schema starten vanaf de dag dat de gemiste dosis is toegediend.
De geplande doseerdag veranderen
Indien patiënten hun geplande doseerdag willen veranderen, kan een nieuwe dosis worden toegediend op de nieuwe gewenste dag van de week. Daarna kan de patiënt vanaf die dag doorgaan met het nieuwe schema, zelf als het doseerinterval tijdelijk korter is dan gebruikelijk.
Overgang van intraveneuze naar subcutane toediening
SLE
Indien een patiënt met SLE wordt overgezet van de intraveneuze toediening van Benlysta naar de subcutane toediening moet de eerste subcutane injectie 1 tot 4 weken na de laatste intraveneuze dosis worden toegediend (zie rubriek 5.2).
Lupusnefritis
Indien een patiënt met lupusnefritis wordt overgezet van intraveneuze toediening van Benlysta naar subcutane toediening, wordt aanbevolen om de eerste subcutane injectiedosis van 200 mg 1 tot 2 weken na de laatste intraveneuze dosis toe te dienen. Deze overgang kan plaatsvinden op enig moment nadat de patiënt de eerste 2 intraveneuze doses heeft voltooid (zie rubriek 5.2).
Speciale patiëntengroepen
Ouderen
Gegevens bij patiënten ≥ 65 jaar zijn beperkt (zie rubriek 5.1). Benlysta dient met voorzichtigheid gebruikt te worden bij ouderen. Een dosisaanpassing is niet vereist (zie rubriek 5.2).
Verminderde nierfunctie
Belimumab is bij een beperkt aantal SLE-patiënten met een verminderde nierfunctie onderzocht. Op basis van de beschikbare informatie is dosisaanpassing niet vereist bij patiënten met een licht, matig of ernstig verminderde nierfunctie. Voorzichtigheid is echter geboden bij patiënten met een ernstig verminderde nierfunctie vanwege het gebrek aan gegevens (zie rubriek 5.2).
Verminderde leverfunctie
Er zijn geen specifieke onderzoeken met Benlysta uitgevoerd bij patiënten met verminderde leverfunctie. Het is onwaarschijnlijk dat bij patiënten met een verminderde leverfunctie een dosisaanpassing vereist is (zie rubriek 5.2).
Pediatrische patiënten
SLE
De veiligheid en werkzaamheid van subcutaan toegediend Benlysta bij kinderen jonger dan 5 jaar of die minder dan 15 kg wegen zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Lupusnefritis
De veiligheid en werkzaamheid van subcutaan toegediend Benlysta bij kinderen en adolescenten jonger dan 18 jaar zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Wijze van toediening
De voorgevulde pen mag alleen worden gebruikt voor subcutane injectie. De aanbevolen injectieplaatsen zijn de buik of de dij. Indien patiënten in dezelfde regio injecteren moet hen worden geadviseerd om voor elke injectie een andere injectieplaats te gebruiken; injecties mogen niet worden gegeven op plaatsen waar de huid gevoelig, gekneusd, rood of hard is. Wanneer een 400 mg dosis wordt toegediend op dezelfde plaats, wordt aanbevolen om de 2 afzonderlijke 200 mg injecties ten minste 5 cm van elkaar toe te dienen.
Begrijpelijke instructies voor subcutane toediening van Benlysta in een voorgevulde pen worden onderaan de bijsluiter gegeven (zie Stapsgewijze instructies).
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De veiligheid van belimumab bij patiënten met SLE is geëvalueerd in drie placebogecontroleerde intraveneuze onderzoeken vóór de registratie en één daaropvolgend regionaal placebogecontroleerd intraveneus onderzoek, één placebogecontroleerd subcutaan onderzoek en twee placebogecontroleerde intraveneuze postmarketingonderzoeken; de veiligheid bij patiënten met actieve lupusnefritis is geëvalueerd in één placebogecontroleerd intraveneus onderzoek.
De in de tabel hieronder gepresenteerde gegevens geven de blootstelling weer bij 674 patiënten met SLE uit de drie klinische onderzoeken vóór de registratie en 470 patiënten uit het daaropvolgende placebogecontroleerde onderzoek die Benlysta intraveneus kregen toegediend (10 mg/kg lichaamsgewicht gedurende een 1 uur durende periode op dagen 0, 14, 28 en dan iedere 28 dagen tot maximaal 52 weken) en 556 patiënten met SLE die werden blootgesteld aan Benlysta subcutaan (maximaal 52 weken eenmaal per week 200 mg). Bij enige patiënten met SLE strekken deze veiligheidsgegevens zich uit over een periode van meer dan 52 weken. De gegevens geven de aanvullende blootstelling weer bij 224 patiënten met actieve lupusnefritis die Benlysta intraveneus kregen (10 mg/kg lichaamsgewicht gedurende maximaal 104 weken). Gegevens uit postmarketingrapportages zijn ook bijgevoegd.
Het merendeel van de patiënten kreeg eveneens een of meer van de volgende gelijktijdige behandelingen voor SLE: corticosteroïden, immunomodulerende geneesmiddelen, antimalariamiddelen, niet-steroïde ontstekingsremmende geneesmiddelen.
Bijwerkingen werden gemeld bij 84 % van de met Benlysta behandelde patiënten en bij 87 % van de met placebo behandelde patiënten. De meest frequent gemelde bijwerking (≥ 5% van de patiënten met SLE die met Benlysta behandeld werden terwijl ze ook een standaardbehandeling kregen en bij een percentage ≥ 1% hoger dan placebo) was nasofaryngitis. Bij de met Benlysta behandelde patiënten stopte 7 % met de behandeling als gevolg van bijwerkingen en bij de met placebo behandelde patiënten 8 %.
De meest frequent gemelde bijwerkingen (> 5% van de patiënten met actieve lupusnefritis die met Benlysta behandeld werden terwijl ze ook een standaardbehandeling kregen) waren luchtweginfecties van de bovenste luchtwegen, urineweginfecties en herpes zoster. Van de met Benlysta behandelde patiënten stopte 12,9 % met de behandeling als gevolg van bijwerkingen en van de met placebo behandelde patiënten 12,9 %.
Ernstige bijwerkingen van de huid: Stevens-Johnson-syndroom (SJS) en toxische epidermale necrolyse (TEN) zijn gemeld in verband met behandeling met Benlysta (zie rubriek 4.4).
Lijst van bijwerkingen in tabelvorm
Hieronder zijn de bijwerkingen gerangschikt volgens de MedDRA-systeem/orgaanklassen en naar frequentie. De frequenties zijn gedefinieerd als:
zeer vaak ≥ 1/10
vaak ≥ 1/100, < 1/10
soms ≥ 1/1.000, < 1/100
zelden ≥ 1/10.000, < 1/1.000
niet bekend kan met de beschikbare gegevens niet worden bepaald.
Binnen iedere frequentiegroep worden bijwerkingen gerangschikt naar afnemende ernst. De frequentie die wordt weergegeven is de hoogste die is gezien, bij ongeacht welke formulering.
Systeem/orgaanklasse | Frequentie | Bijwerkingen |
Infecties en parasitaire aandoeningen1 | Zeer vaak | Bacteriële infecties, bijvoorbeeld bronchitis, urineweginfectie |
Vaak | Virale gastro-enteritis, faryngitis, nasofaryngitis, virale luchtweginfectie van de bovenste luchtwegen | |
Bloed- en lymfestelselaandoeningen | Vaak | Leukopenie |
Immuunsysteemaandoeningen | Vaak | Overgevoeligheidsreacties2 |
Soms | Anafylactische reactie | |
Zelden | Niet-acute overgevoeligheidsreacties van het vertraagde type | |
Psychische stoornissen | Vaak | Depressie |
Soms | Suïcidaal gedrag, suïcidale gedachten | |
Zenuwstelselaandoeningen | Vaak | Migraine |
Maagdarmstelselaandoeningen | Vaak | Diarree, misselijkheid |
Huid- en onderhuidaandoeningen | Vaak | Injectieplaatsreacties3, urticaria, rash |
Soms | Angio-oedeem | |
Niet bekend | Stevens-Johnson-syndroom, toxische epidermale necrolyse | |
Skeletspierstelsel- en bindweefselaandoeningen | Vaak | Pijn in extremiteit |
Algemene aandoeningen en toedieningsplaatsstoornissen | Vaak | Infuus- of injectiegerelateerde systemische reacties2, koorts |
1 Zie ‘Beschrijving van geselecteerde bijwerkingen’ en rubriek 4.4 ‘Infecties’ voor verder informatie.
2 ‘Overgevoeligheidsreacties’ is een verzamelnaam voor een groep begrippen, waaronder anafylaxie, die zich kunnen openbaren met een scala aan symptomen zoals hypotensie, angio-oedeem, urticaria of andere vormen van rash, pruritus en dyspneu. ‘Infuus- of injectiegerelateerde systemische reacties’ is een verzamelnaam voor een groep begrippen die zich kunnen openbaren met een scala aan symptomen zoals bradycardie, myalgie, hoofdpijn, rash, urticaria, koorts, hypotensie, hypertensie, duizeligheid en artralgie. Omdat er een overlapping is wat betreft klachten en symptomen is het niet mogelijk om in alle gevallen onderscheid te maken tussen overgevoeligheidsreacties en infuus- of injectiegerelateerde systemische reacties.
3 Dit is alleen van toepassing bij de subcutane formulering.
Beschrijving van geselecteerde bijwerkingen
Gegevens die hieronder worden gepresenteerd zijn gegroepeerd uit de drie intraveneuze klinische onderzoeken vóór de registratie (alleen de 10 mg/kg lichaamsgewicht intraveneuze dosering) en het subcutane klinische onderzoek. Bij de ‘Infecties’ en ‘Psychische stoornissen’ zijn ook gegevens opgenomen uit een postmarketingonderzoek.
Infuus- of injectiegerelateerde systemische reacties en overgevoeligheid: infuus- of injectie-gerelateerde systemische reacties en overgevoeligheidsreacties werden over het algemeen waargenomen op de dag van toediening, maar acute overgevoeligheidsreacties kunnen ook plaatsvinden enkele dagen na de toediening. Patiënten bij wie in het verleden meerdere geneesmiddelallergieën of significante overgevoeligheidsreacties zijn opgetreden, kunnen een verhoogd risico lopen.
De incidentie van infuusreacties en overgevoeligheidsreacties na intraveneuze toediening die optreedt binnen 3 dagen na toediening van een infusie was 12 % bij de groep die Benlysta kreeg en 10% in de placebogroep. Hierbij moesten respectievelijk 1,2 % en 0,3 % van de patiënten definitief met de behandeling stoppen.
De incidentie van systemische en overgevoeligheidsreacties na injectie die binnen 3 dagen na subcutane toediening optraden was 7 % in de groep die Benlysta kreeg en 9 % in de groep die placebo kreeg. Klinisch significante overgevoeligheidsreacties die in verband werden gebracht met subcutaan toegediende Benlysta en waarvoor permanent staken van de behandeling noodzakelijk was, werd gemeld bij 0, 2% van de patiënten die Benlysta kreeg en bij geen patiënten die placebo kregen.
Infecties: de totale infectie-incidentie bij intraveneuze en subcutane pre-registratie SLE-studies was 63 % in de beide groepen die Benlysta of placebo kregen. Infecties die bij ten minste 3% van de Benlysta-patiënten en ten minste 1 % vaker dan bij de placebopatiënten optraden, waren virale luchtweginfecties van de bovenste luchtwegen, bronchitis en bacteriële urineweginfecties. Ernstige infecties traden op bij 5 % van de patiënten in beide groepen die Benlysta of placebo kregen; ernstige opportunistische infecties bij respectievelijk 0,4 % en 0 % hiervan. Bij 0,7 % van de Benlysta-patiënten en bij 1,5 % van de placebopatiënten leidden infecties tot stopzetting van de behandeling. Sommige infecties waren ernstig of fataal.
Voor informatie over waargenomen infecties bij pediatrische patiënten met SLE, zie ‘Pediatrische patiënten’ hieronder.
In het lupusnefritis-onderzoek kregen patiënten een standaardbehandeling als achtergrondbehandeling (zie rubriek 5.1) en was de totale infectie‑incidentie 82 % bij patiënten die Benlysta kregen in vergelijking met 76 % bij patiënten die placebo kregen. Ernstige infecties traden op bij 13,8 % van de patiënten die Benlysta kregen en bij 17,0 % van de patiënten die placebo kregen. Fatale infecties traden op bij 0,9 % (2/224) van de patiënten die Benlysta kregen en bij 0,9 % (2/224) van de patiënten die placebo kregen.
In een gerandomiseerd, dubbelblind, 52 weken durend postmarketing SLE-veiligheidsonderzoek (BEL115467) waarin mortaliteit en specifieke bijwerkingen bij volwassenen werden beoordeeld, traden ernstige infecties op bij 3,7 % van de patiënten die Benlysta 10 mg/kg lichaamsgewicht intraveneus kregen en bij 4,1 % van de patiënten die placebo kregen. Fatale infecties (zoals longontsteking en sepsis) traden echter op bij 0,4 5% (9/2002) van de patiënten die Benlysta kregen versus 0,15 % (3/2001) van de patiënten die placebo kregen, terwijl de incidentie van mortaliteit door alle oorzaken respectievelijk 0,50 % (10/2002) was versus 0,40 % (8/2001). De meeste fatale infecties werden gedurende de eerste 20 weken van de behandeling met Benlysta gezien.
Psychische stoornissen: in de intraveneuze klinische SLE-onderzoeken vóór de registratie werden ernstige psychische voorvallen gemeld bij 1,2 % (8/674) van de patiënten die Benlysta 10 mg/kg lichaamsgewicht kregen en bij 0,4 % (3/675) van de patiënten die placebo kregen. Ernstige depressie werd gemeld bij 0,6 % (4/674) van de patiënten die Benlysta 10 mg/kg lichaamsgewicht kregen en bij 0,3 % (2/675) van de patiënten die placebo kregen. Er waren twee gevallen van suïcide in de groep patiënten die met Benlysta werd behandeld (onder wie een patiënt die Benlysta 1 mg/kg lichaamsgewicht kreeg).
In een postmarketing SLE-onderzoek werden ernstige psychische voorvallen gemeld bij 1,0 % (20/2002) van de patiënten die Benlysta kregen en bij 0,3 % (6/2001) van de patiënten die placebo kregen. Ernstige depressie werd gemeld bij 0,3 % (7/2002) van de patiënten die Benlysta kregen en bij < 0, 1% (1/2001) van de patiënten die placebo kregen. De totale incidentie van ernstige suïcidale gedachten of suïcidaal gedrag of automutilatie zonder suïcidale intentie was 0,7 % (15/2002) bij patiënten die Benlysta kregen en 0,2 % (5/2001) in de placebogroep. In geen van de groepen werd suïcide gemeld.
In de bovenstaande intraveneuze SLE-onderzoeken werden patiënten met een voorgeschiedenis van psychische stoornissen niet uitgesloten.
In het subcutane klinische SLE-onderzoek, waarin patiënten met een voorgeschiedenis van psychische stoornissen werden uitgesloten, werden ernstige psychische voorvallen gemeld bij 0,2 % (1/556) van de patiënten die Benlysta kregen en bij geen van de patiënten die placebo kregen. In geen van de groepen werden voorvallen vanwege ernstige depressie of gevallen van suïcide gemeld.
Leukopenie: de incidentie van als bijwerking gemelde leukopenie bij patiënten met SLE was 3 % in de Benlystagroep en 2 % in de placebogroep.
Injectieplaatsreacties: in de subcutane SLE-studie was de frequentie van injectieplaatsreacties respectievelijk 6,1 % (34/556) en 2,5 % (7/280) voor patiënten die Benlysta of placebo kregen. Deze injectieplaatsreacties (het vaakste pijn, erytheem, hematoom, pruritus en induratie) waren licht tot matig van ernst. Voor de meeste reacties was het niet nodig het gebruik van het geneesmiddel te staken.
Pediatrische patiënten
Het bijwerkingenprofiel bij pediatrische patiënten is gebaseerd op één subcutaan onderzoek en één intraveneus onderzoek.
In een 52 weken durend open-labelonderzoek waarbij 25 pediatrische patiënten (van 10 tot 17 jaar oud) met SLE subcutaan Benlysta kregen toegediend met een blootstelling die vergelijkbaar was met die bij volwassenen (200 mg met een vast doseringsinterval op basis van lichaamsgewicht, tegen een achtergrond van gelijktijdige behandelingen), was het veiligheidsprofiel bij pediatrische patiënten die subcutaan Benlysta toegediend kregen, vergelijkbaar met het bekende veiligheidsprofiel van belimumab.
In een 52 weken durend placebogecontroleerd onderzoek waarbij 53 patiënten (van 6 tot 17 jaar oud) met SLE Benlysta kregen toegediend (10 mg/kg lichaamsgewicht intraveneus op dag 0, 14 en 28 en vervolgens elke 28 dagen, tegen een achtergrond van gelijktijdige behandelingen), werden geen nieuwe veiligheidssignalen gezien bij pediatrische patiënten van 12 jaar en ouder (n = 43). De veiligheidsgegevens bij kinderen jonger dan 12 jaar (n = 10) zijn beperkt.
Infecties
Groep met 5- tot 11-jarigen: infecties werden gemeld bij 8/10 patiënten die intraveneus Benlysta toegediend kregen en 3/3 patiënten die placebo ontvingen, en ernstige infecties werden gemeld bij 1/10 patiënten die intraveneus Benlysta toegediend kregen en 2/3 patiënten die placebo kregen (zie rubriek 4.4).
Groep met 12- tot 17-jarigen: infecties werden gemeld bij 22/43 patiënten die intraveneus Benlysta toegediend kregen en 25/37 patiënten die placebo kregen, en ernstige infecties werden gemeld bij 3/43 patiënten die intraveneus Benlysta toegediend kregen en 3/37 patiënten die placebo kregen. In de open-label extensiefase was er één fatale infectie bij een patiënt die Benlysta intraveneus toegediend kreeg.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem:
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be

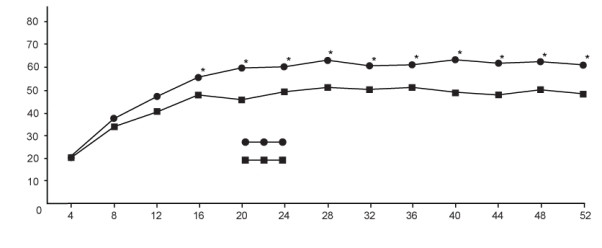
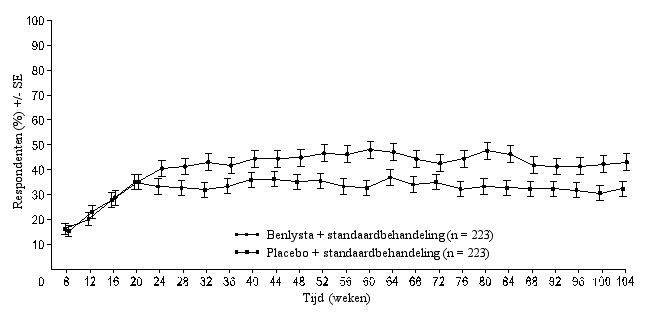 ‘Primary Efficacy Renal Response’ (PERR)
‘Primary Efficacy Renal Response’ (PERR)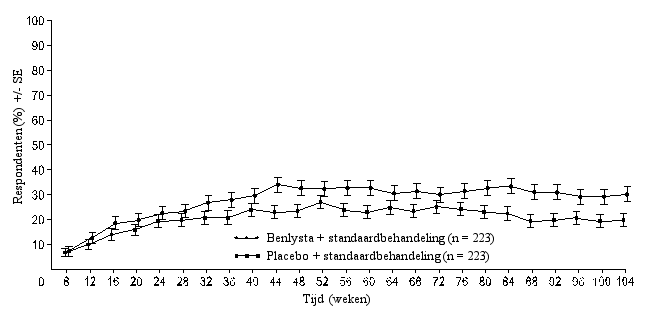 ‘Complete Renal Response’ (CRR)
‘Complete Renal Response’ (CRR)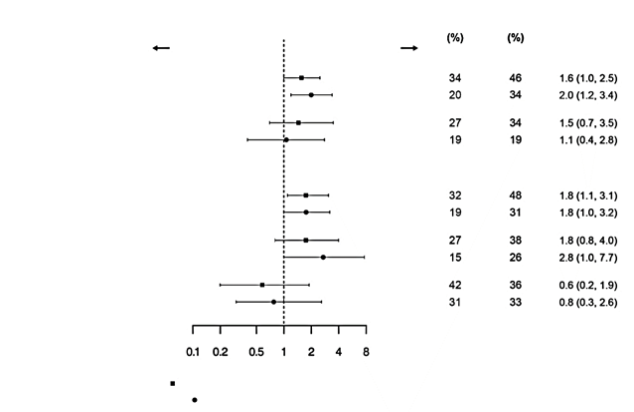
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
GlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Ierland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/11/700/003 1 voorgevulde pen
EU/1/11/700/004 4 voorgevulde pennen
EU/1/11/700/005 12 (3x4) voorgevulde pennen (multiverpakking)
10. DATUM VAN HERZIENING VAN DE TEKST
18/07/2025
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu/.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3716115 | BENLYSTA 200MG OPL INJ VOORGEVULDE PEN 4 X 1ML | L04AA26 | - | - | Ja | - | - |