SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Alecensa 150 mg harde capsules
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke harde capsule bevat alectinibhydrochloride overeenkomend met 150 mg alectinib.
Hulpstoffen met bekend effect:
Elke harde capsule bevat 33,7 mg lactose (als monohydraat) en 6 mg natrium (als natriumlaurylsulfaat).
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Harde capsule.
Witte harde capsule met een lengte van 19,2 mm, bedrukt met “ALE” in zwarte inkt op de dop en bedrukt met “150 mg” in zwarte inkt op de romp.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
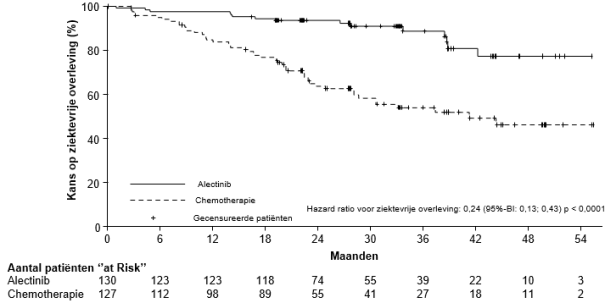
Adjuvante behandeling van gereseceerde niet-kleincellige longkanker (NSCLC)
Alecensa is als monotherapie geïndiceerd als adjuvante behandeling na volledige tumorresectie voor volwassen patiënten met ALK-positieve NSCLC met een hoog risico op recidief (zie rubriek 5.1 voor de selectiecriteria).
Behandeling van gevorderde NSCLC
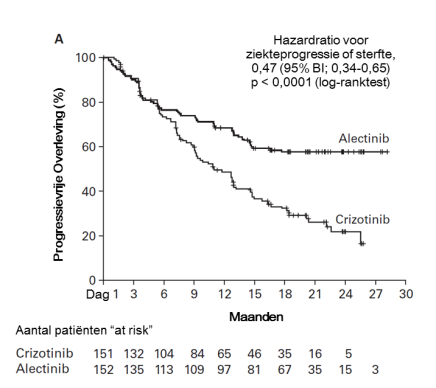
Alecensa is als monotherapie geïndiceerd voor de eerstelijnsbehandeling van volwassen patiënten met ALK-positieve gevorderde NSCLC.
Alecensa is als monotherapie geïndiceerd voor de behandeling van volwassen patiënten met ALK-positieve gevorderde NSCLC, die eerder behandeld zijn met crizotinib.
4.2 Dosering en wijze van toediening
De behandeling met Alecensa moet worden gestart onder toezicht van een arts met ervaring in het gebruik van geneesmiddelen tegen kanker.
Een gevalideerde ALK-test is noodzakelijk voor het selecteren van patiënten met ALK-positieve NSCLC. NSCLC met een ALK-positieve status moet worden vastgesteld vóór de aanvang van de behandeling met Alecensa.
Dosering
De aanbevolen dosering van Alecensa is 600 mg (4 capsules van 150 mg) tweemaal daags ingenomen met voedsel (totale dagelijkse dosis van 1.200 mg).
Patiënten met een onderliggend ernstig verminderde leverfunctie (Child-Pugh C) moeten een aanvangsdosering krijgen van 450 mg tweemaal daags ingenomen met voedsel (totale dagelijkse dosis van 900 mg).
Duur van de behandeling
Adjuvante behandeling van gereseceerde NSCLC
Behandeling met Alecensa moet worden voortgezet tot terugkeer van ziekte, onaanvaardbare toxiciteit of gedurende 2 jaar.
Behandeling van gevorderde NSCLC
Behandeling met Alecensa moet worden voortgezet tot ziekteprogressie of onaanvaardbare toxiciteit.
Uitgestelde of gemiste doses
Wanneer een geplande dosis van Alecensa wordt gemist, kunnen patiënten die dosis innemen, tenzij het minder dan 6 uur is tot de volgende dosis. Patiënten moeten geen dubbele dosis innemen om een gemiste dosis in te halen. In het geval van braken na het innemen van een dosis van Alecensa moeten patiënten de volgende dosis op het geplande tijdstip innemen.
Doseringsaanpassingen
In het geval van bijwerkingen kan het nodig zijn om de dosering te verlagen, tijdelijk te onderbreken of de behandeling met Alecensa te beëindigen. De dosering van Alecensa moet in stappen van 150 mg tweemaal daags worden verlaagd op basis van verdraagbaarheid. De behandeling met Alecensa moet permanent worden beëindigd als patiënten niet in staat zijn om de tweemaal daagse dosering van 300 mg te verdragen.
Advies over doseringsaanpassing staat hieronder vermeld in tabel 1 en 2.
Tabel 1 Schema voor doseringsverlaging
Schema voor doseringsverlaging | Dosering |
Dosering | 600 mg tweemaal daags |
Eerste doseringsverlaging | 450 mg tweemaal daags |
Tweede doseringsverlaging | 300 mg tweemaal daags |
Tabel 2 Advies over doseringsaanpassing bij bepaalde bijwerkingen (zie rubriek 4.4 en 4.8)
CTC-AE graad | Behandeling met Alecensa |
ILD / pneumonitis van elke graad | Onmiddellijk de behandeling onderbreken en definitief de behandeling met Alecensa beëindigen als er geen andere mogelijke oorzaken van ILD / pneumonitis zijn vastgesteld. |
ALAT- of ASAT-verhoging > 5 keer ULN met totaal bilirubine ≤ 2 keer ULN | Tijdelijk de behandeling onderbreken tot verbetering naar baseline of ≤ 3 keer ULN, hervat met een lagere dosering (zie tabel 1). |
ALAT- of ASAT-verhoging > 3 keer ULN met verhoging van totaal bilirubine > 2 keer ULN in afwezigheid van cholestase of hemolyse | Definitief de behandeling met Alecensa beëindigen. |
Graad 2 of graad 3 bradycardiea (symptomatisch, kan ernstig en medisch significant zijn, medische interventie geïndiceerd) | Tijdelijk de behandeling onderbreken tot verbetering naar graad ≤ 1 (asymptomatische) bradycardie of tot een hartslag van ≥ 60 bpm. Evalueer co-medicatie waarvan bekend is dat het bradycardie veroorzaakt, alsmede antihypertensiva. |
Graad 4 bradycardiea (levensbedreigende gevolgen, dringende interventie geïndiceerd) | Definitief de behandeling met Alecensa beëindigen als er geen bijdragende co-medicatie is vastgesteld. |
CPK-verhoging > 5 keer ULN | Tijdelijk de behandeling onderbreken tot verbetering naar baseline of tot ≤ 2,5 keer ULN, hervat met dezelfde dosering. |
CPK-verhoging > 10 keer ULN of | Tijdelijk de behandeling onderbreken tot verbetering naar baseline of tot ≤ 2,5 keer ULN, hervat met een lagere dosering (zie tabel 1). |
Hemolytische anemie met hemoglobine < 10 g/dl (graad ≥ 2) | Tijdelijk de behandeling onderbreken tot herstel, vervolgens hervatten met een lagere dosering (zie tabel 1). |
ALAT = alanineaminotransferase; ASAT = aspartaataminotransferase; CPK = creatinefosfokinase; CTC‑AE = NCI Common Terminology Criteria for Adverse Events; ILD = interstitiële longziekte; ULN = bovenlimiet van de normaalwaarde.
a Hartslag minder dan 60 slagen per minuut (bpm).
Speciale populaties
Verminderde leverfunctie
Er is geen aanpassing van de aanvangsdosering nodig bij patiënten met een onderliggend licht (Child‑Pugh A) of matig (Child‑Pugh B) verminderde leverfunctie. Patiënten met een onderliggend ernstig (Child‑Pugh C) verminderde leverfunctie moeten een aanvangsdosering van 450 mg tweemaal daags krijgen (totale dosis van 900 mg) (zie rubriek 5.2). Gepaste controle (bijv. leverfunctiewaarden) wordt aangeraden voor alle patiënten met een verminderde leverfunctie; zie rubriek 4.4.
Verminderde nierfunctie
Er is geen doseringsaanpassing nodig bij patiënten met een licht of matig verminderde nierfunctie. Alecensa is niet onderzocht bij patiënten met een ernstig verminderde nierfunctie. Aangezien de eliminatie van alectinib via de nieren echter verwaarloosbaar is, is er geen doseringsaanpassing vereist bij patiënten met een ernstig verminderde nierfunctie (zie rubriek 5.2).
Ouderen (≥ 65 jaar)
De beperkte gegevens over de veiligheid en werkzaamheid van Alecensa bij patiënten van 65 jaar en ouder wijzen er niet op dat een doseringsaanpassing bij oudere patiënten nodig is (zie rubriek 5.2). Er zijn geen gegevens beschikbaar over patiënten ouder dan 80 jaar.
Pediatrische patiënten
De veiligheid en werkzaamheid van Alecensa bij kinderen en adolescenten jonger dan 18 jaar zijn niet vastgesteld. Er zijn geen gegevens beschikbaar.
Extreem lichaamsgewicht (> 130 kg)
Hoewel farmacokinetische (PK) simulaties met Alecensa geen lage blootstelling aantonen bij patiënten met extreem lichaamsgewicht (d.w.z. > 130 kg), wordt alectinib door het hele lichaam verspreid en het lichaamsgewicht van patiënten die deelnamen aan klinische studies met alectinib varieerde van 36,9 - 123 kg. Er zijn geen gegevens beschikbaar over patiënten met een lichaamsgewicht boven de 130 kg.
Wijze van toediening
Alecensa is voor oraal gebruik. De harde capsules moeten in hun geheel worden doorgeslikt. Ze mogen niet worden geopend of worden opgelost. Ze dienen met voedsel te worden ingenomen (zie rubriek 5.2).
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De hieronder beschreven gegevens geven de blootstelling weer van Alecensa bij 533 patiënten met gereseceerde of gevorderde ALK-positieve NSCLC. Deze patiënten kregen Alecensa in de aanbevolen dosering van 600 mg tweemaal daags in klinische registratieonderzoeken voor adjuvante behandeling van gereseceerde NSCLC (BO40336, ALINA) of voor de behandeling van gevorderde NSCLC (BO28984, ALEX; NP28761; NP28673). Zie rubriek 5.1 voor meer informatie over de deelnemers aan de klinische onderzoeken.
In BO40336 (ALINA; N = 128) was de mediane blootstellingsduur aan Alecensa 23,9 maanden. In BO28984 (ALEX; N = 152) was de mediane blootstellingsduur aan Alecensa 28,1 maanden. In de klinische fase II-onderzoeken (NP28761, NP28673; N = 253) was de mediane blootstellingsduur aan Alecensa 11,2 maanden.
De meest voorkomende bijwerkingen (≥ 20%) waren constipatie, myalgie, oedeem, verhoogde bilirubine, verhoogde ASAT, anemie, huiduitslag en verhoogde ALAT.
Tabel met een samenvatting van de bijwerkingen
Tabel 3 is een samenvatting van bijwerkingen die optraden bij patiënten die Alecensa kregen in klinische onderzoeken (BO40336, BO28984, NP28761, NP28673).
De bijwerkingen in tabel 3 worden weergegeven per systeem/orgaanklasse en frequentiecategorie, gedefinieerd als: zeer vaak (≥ 1/10), vaak (≥ 1/100 tot < 1/10), soms (≥ 1/1.000 tot < 1/100), zelden (≥ 1/10.000 tot < 1/1.000), zeer zelden (< 1/10.000). Binnen elke systeem/orgaanklasse zijn de bijwerkingen gerangschikt in volgorde van afnemende frequentie en graad van ernst. Binnen dezelfde frequentiecategorie en ernstgradering worden bijwerkingen weergegeven in volgorde van afnemende ernst.
Tabel 3 Bijwerkingen gemeld in klinische onderzoeken met Alecensa (BO40336, BO28984, NP28761, NP28673; N = 533)
Systeem/orgaanklasse | Alecensa | |
| Frequentiecategorie | Frequentiecategorie |
Bloed- en lymfestelselaandoeningen | ||
Anemie1) | Zeer vaak | Vaak |
Hemolytische anemie2) | Vaak | -* |
Zenuwstelselaandoeningen | ||
Dysgeusie3) | Vaak | Soms |
Oogaandoeningen | ||
Visusstoornissen4) | Vaak | -* |
Hartaandoeningen | ||
Bradycardie5) | Zeer vaak | -* |
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen | ||
Interstitiële longziekte (ILD)/ pneumonitis | Vaak | Soms |
Maagdarmstelselaandoeningen | ||
Diarree | Zeer vaak | Vaak |
Braken | Zeer vaak | Soms |
Constipatie | Zeer vaak | Soms |
Misselijkheid | Zeer vaak | Soms |
Stomatitis6) | Vaak | Soms |
Lever- en galaandoeningen | ||
Verhoogde ASAT | Zeer vaak | Vaak |
Verhoogde ALAT | Zeer vaak | Vaak |
Verhoogde bilirubine7) | Zeer vaak | Vaak |
Verhoogde alkalische fosfatase | Zeer vaak | Soms |
Geneesmiddelgeïnduceerd leverletsel8) | Soms | Soms |
Huid- en onderhuidaandoeningen | ||
Huiduitslag9) | Zeer vaak | Vaak |
Lichtgevoeligheid | Vaak | Soms |
Skeletspierstelsel- en bindweefselaandoeningen | ||
Myalgie10) | Zeer vaak | Soms |
Verhoogde creatine fosfokinase (CPK) in het bloed | Zeer vaak | Vaak |
Nier- en urinewegaandoeningen | ||
Verhoogde creatinine in het bloed | Zeer vaak | Soms** |
Acuut nierletsel | Vaak | Soms** |
Algemene aandoeningen en toedieningsplaatsstoornissen | ||
Oedeem11) | Zeer vaak | Soms |
Onderzoeken | ||
Gewichtstoename | Zeer vaak | Soms |
Voedings- en stofwisselingsstoornissen | ||
Hyperurikemie12) | Vaak | -* |
*er werden geen bijwerkingen van graad 3-4 waargenomen.
**waaronder één voorval van graad 5 (waargenomen in de gevorderde NSCLC-setting).
1) waaronder gevallen van anemie, verlaagd hemoglobine en normochrome normocytaire anemie.
2) gevallen gemeld in onderzoek BO40336 (N = 128).
3) waaronder gevallen van dysgeusie, hypogeusie en smaakstoornis.
4) waaronder gevallen van wazig zien, slechter zien, mouches volantes, verminderde gezichtsscherpte, asthenopie, dubbelzien, fotofobie en fotopsie.
5) waaronder gevallen van bradycardie en sinusbradycardie.
6) waaronder gevallen van stomatitis en mondzweren.
7) waaronder gevallen van verhoogde bloedbilirubine, hyperbilirubinemie, verhoogde geconjugeerde bilirubine en verhoogde niet-geconjungeerde bloedbilirubine.
8) waaronder twee patiënten waarbij geneesmiddelgeïnduceerd leverletsel is gerapporteerd volgens MedDRA terminologie en één patiënt waarbij verhoogde ASAT/ALAT van graad 4 is gemeld en bij wie geneesmiddelgeïnduceerd leverletsel is vastgesteld door middel van leverbiopsie.
9) waaronder gevallen van huiduitslag, maculopapulaire huiduitslag, dermatitis, acneïforme dermatitis, erytheem, papulaire huiduitslag, pruritische huiduitslag, maculaire huiduitslag, exfoliatieve huiduitslag en erythemateuze huiduitslag.
10) waaronder gevallen van myalgie, musculoskeletale pijn en artralgie.
11) waaronder gevallen van perifeer oedeem, oedeem, gegeneraliseerd oedeem, ooglidoedeem, periorbitaal oedeem, gezichtsoedeem, gelokaliseerd oedeem, perifere zwelling, zwelling van gezicht, lipzwelling, zwelling, gewrichtszwelling en zwelling van ooglid.
12) waaronder gevallen van hyperurikemie en verhoogde urinezuurwaarden in het bloed.
Beschrijving van geselecteerde bijwerkingen
Interstitiële longziekte (ILD)/pneumonitis
In de klinische onderzoeken kwam ILD/pneumonitis bij 1,7% van de patiënten voor die met Alecensa werden behandeld. In 0,4% van deze gevallen was dit graad 3 en beëindiging van de behandeling vanwege ILD/pneumonitis kwam bij 1,1% van de patiënten voor en bij 0,4% van de patiënten leidde het voorval tot doseringsaanpassingen. In het klinische fase III-onderzoek BO28984 werd er geen graad 3 of graad 4 ILD/pneumonitis waargenomen bij patiënten die Alecensa kregen versus 2,0% bij patiënten die crizotinib kregen. In de klinische onderzoeken waren er geen fatale gevallen van ILD. Patiënten moeten worden gecontroleerd op longsymptomen die wijzen op pneumonitis (zie rubriek 4.2 en 4.4).
Hepatotoxiciteit
In de klinische onderzoeken werd bij drie patiënten geneesmiddelgeïnduceerd leverletsel vastgesteld (waaronder twee patiënten met de gemelde term geneesmiddelgeïnduceerd leverletsel en één patiënt met gemelde graad 4 ASAT/ALAT-verhogingen, bij wie geneesmiddelgeïnduceerd leverletsel werd vastgesteld door middel van leverbiopsie). Bijwerkingen van verhoogde ASAT- en ALAT-waarden (respectievelijk 23,6% en 20,5%) werden gemeld bij patiënten die met Alecensa werden behandeld in de klinische onderzoeken. De meerderheid van deze gevallen waren graad 1 en 2; gevallen van graad ≥ 3 werden gemeld bij respectievelijk 3,0% en 3,2% van de patiënten voor toegenomen ASAT- en ALAT-waarden. De gevallen traden meestal op tijdens de eerste 3 maanden van de behandeling en waren gewoonlijk van voorbijgaande aard en verdwenen na tijdelijke onderbreking van de Alecensa-behandeling (gemeld bij respectievelijk 2,3% en 3,6% van de patiënten) of na een doseringsverlaging (respectievelijk 1,7% en 1,5%). Bij 1,3% en 1,5% van de patiënten leidden de verhogingen in respectievelijk ASAT en ALAT tot beëindiging van de Alecensa-behandeling. Graad 3 of graad 4 ALAT/ASAT-verhogingen werden waargenomen bij 4,6% en 5,3% van de patiënten die Alecensa kregen versus 16,6% en 10,6% van patiënten die crizotinib kregen in het klinische fase III-onderzoek BO28984.
In de klinische onderzoeken werden bijwerkingen van verhoogde bilirubine gemeld bij 25,9% van de patiënten die met Alecensa werden behandeld. De meerderheid van de gevallen waren graad 1 en 2; gevallen van graad ≥ 3 werden bij 3,9% van de patiënten gemeld. De gevallen traden meestal op tijdens de eerste 3 maanden van de behandeling, waren gewoonlijk van voorbijgaande aard en de meerderheid verdween na doseringsaanpassing. Bij 8,3% van de patiënten leidden de verhogingen van bilirubine tot doseringsaanpassingen en bij 2,1% van de patiënten leidden de verhogingen van bilirubine tot beëindiging van de Alecensa-behandeling. In het klinische fase III-onderzoek BO28984 traden graad 3 of graad 4 verhogingen van bilirubine op bij 5,9% van de patiënten die Alecensa kregen versus bij geen patiënt die crizotinib kreeg.
Gelijktijdige verhogingen van ALAT of ASAT van meer dan of gelijk aan 3 keer de ULN en totale bilirubine hoger dan of gelijk aan 2 keer de ULN, met normale alkalinefosfatase, kwam voor bij 1 patiënt (0,2%) die werd behandeld met Alecensa in klinische onderzoeken.
Patiënten moeten worden gecontroleerd op leverfunctie, waaronder ALAT, ASAT en totale bilirubine, zoals uiteengezet in rubriek 4.4 en behandeld zoals geadviseerd in rubriek 4.2.
Bradycardie
Gevallen van bradycardie (11,3%) van graad 1 of 2 zijn in de klinische onderzoeken gemeld bij patiënten die werden behandeld met Alecensa. Geen van de patiënten kregen voorvallen van graad ≥ 3. Van de 521 patiënten die met Alecensa werden behandeld, hadden 102 (19,6%), voor wie periodieke ECG’s beschikbaar waren, na de dosering hartslagwaarden onder 50 slagen per minuut (bpm). In het klinische fase III-onderzoek BO28984 had 12,4% van de patiënten die met Alecensa werden behandeld hartslagwaarden onder 50 bpm versus 17,6% van de patiënten die met crizotinib werden behandeld. Patiënten die symptomatische bradycardie ontwikkelen moeten worden behandeld zoals aanbevolen in rubrieken 4.2 en 4.4. Geen van de gevallen van bradycardie leidde tot beëindiging van de Alecensa-behandeling.
Ernstige myalgie en creatinefosfokinase (CPK)-verhoging
Gevallen van myalgie (35,3%), waaronder voorvallen van myalgie (24,2%), artralgie (16,3%) en musculoskeletale pijn (0,8%), zijn in de klinische onderzoeken gemeld bij patiënten die werden behandeld met Alecensa. De meerderheid van de gevallen was graad 1 of 2 en 5 patiënten (0,9%) hadden een voorval van graad 3. Doseringsaanpassingen van de Alecensa-behandeling als gevolg van deze bijwerkingen waren bij 9 patiënten nodig (1,7%); de Alecensa-behandeling werd niet beëindigd door deze gevallen van myalgie. Verhogingen van CPK kwamen voor bij 56,2% van de 491 patiënten voor wie CPK-laboratoriumgegevens beschikbaar waren uit klinische onderzoeken met Alecensa. De incidentie van graad ≥ 3 CPK-verhogingen was 5,5%. De mediane tijd tot graad ≥ 3 CPK-verhoging was 15 dagen in de klinische onderzoeken. Doseringsaanpassingen voor CPK-verhoging kwamen voor bij 5,4% van de patiënten; beëindiging van de Alecensa-behandeling door CPK-verhoging kwam niet voor. In het klinisch onderzoek BO28984 werd ernstige artralgie gemeld bij 1 patiënt (0,7%) in de alectinib-behandelarm en bij 2 patiënten (1,3%) in de crizotinib-behandelarm. Graad ≥ 3 verhoging van CPK werd gemeld bij 3,3% van de patiënten die Alecensa kregen en bij 4,6% van de patiënten die crizotinib kregen.
Hemolytische anemie
Hemolytische anemie werd waargenomen bij 3,1% van de patiënten die werden behandeld met Alecensa in de klinische onderzoeken. De ernst van deze gevallen was graad 1 of 2 (niet ernstig) en leidde niet tot onderbreking van de behandeling (zie rubriek 4.2 en 4.4).
Maagdarmstelselaandoeningen
Constipatie (39,6%), diarree (18,8%), misselijkheid (17,6%) en braken (12,4%) waren de meest vaak gemelde maagdarmstelselreacties. De meeste gevallen waren van milde of matige ernst; graad 3 gevallen werden gemeld voor diarree (1,1%), misselijkheid (0,4%), constipatie (0,4%) en braken (0,2%). Deze gevallen leidden niet tot beëindiging van de Alecensa-behandeling. De mediane tijd tot optreden van de bijwerkingen constipatie, misselijkheid, diarree en/of braken was in de klinische onderzoeken 21 dagen. De frequentie van de bijwerkingen verminderde na de eerste maand van de behandeling. In het klinisch fase III-onderzoek BO28984 werden graad 3 en 4 voorvallen van misselijkheid en constipatie gemeld bij 1 patiënt elk (0,7%), terwijl diarree gemeld werd bij 2 patiënten (1,3%) in de alectinib-behandelarm; de incidentie van graad 3 en 4 voorvallen van misselijkheid, braken en diarree was respectievelijk 3,3%, 3,3% en 2,0%, in de crizotinib-behandelarm.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden (zie hieronder voor details).
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/16/1169/001
EU/1/16/1169/002
10. DATUM VAN HERZIENING VAN DE TEKST
29 januari 2026
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau https://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 3518990 | ALECENSA 150MG HARDE CAPS 4 X 56 | L01ED03 | - | € 4872,14 | Ja | - | - |