SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Zelboraf 240 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke tablet bevat 240 mg vemurafenib (als een gecombineerde neerslag van vemurafenib en hypromelloseacetaatsuccinaat).
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet).
Rozeachtig-witte tot oranje-witte, ovale, biconvexe filmomhulde tabletten van ongeveer 19 mm, met “VEM” op één zijde gegraveerd.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
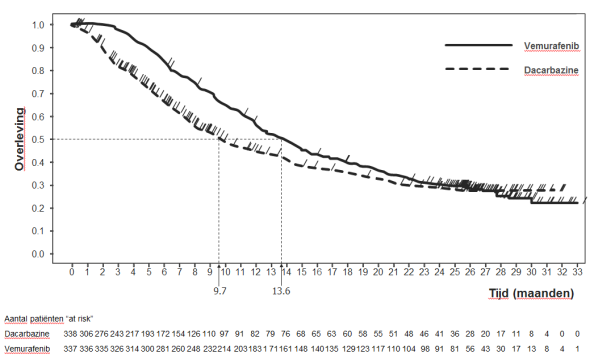
Vemurafenib is geïndiceerd als monotherapie voor de behandeling van volwassen patiënten met een inoperabel of gemetastaseerd melanoom dat positief is voor de BRAF V600-mutatie (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
De behandeling met vemurafenib dient geïnitieerd te worden door en onder toezicht plaats te vinden van een bevoegd arts met ervaring met het gebruik van oncologische geneesmiddelen.
Alvorens vemurafenib te gebruiken, moeten patiënten een bevestiging hebben dat de tumor positief is voor de BRAF V600-mutatie, door middel van een gevalideerde test (zie rubrieken 4.4 en 5.1).
Dosering
De aanbevolen dosering van vemurafenib is 960 mg (4 tabletten van 240 mg) tweemaal daags (gelijk aan een totale dagelijkse dosis van 1.920 mg). Vemurafenib mag zowel met als zonder voedsel worden ingenomen, maar consequente inname van beide dagelijkse doses op een lege maag moet worden vermeden (zie rubriek 5.2).
Duur van de behandeling
Behandeling met vemurafenib moet voortgezet worden tot progressie van de ziekte of het optreden van onacceptabele toxiciteit (zie tabellen 1 en 2 hieronder).
Gemiste doses
Wanneer een dosis wordt gemist, kan deze worden ingenomen tot 4 uur voorafgaand aan de volgende dosis om zo het tweemaal daagse schema te behouden. Beide doses mogen niet gelijktijdig ingenomen worden.
Braken
In geval van braken nadat vemurafenib is ingenomen, dient de patiënt geen extra dosis van het geneesmiddel te nemen, maar de behandeling ongewijzigd voort te zetten.
Doseringsaanpassingen
Voor het behandelen van bijwerkingen of QTc-verlenging kan het nodig zijn om de dosering te verlagen, de toediening tijdelijk te onderbreken en/of de behandeling te staken (zie tabellen 1 en 2). Aanpassing van de dosering tot minder dan 480 mg tweemaal daags wordt niet aanbevolen.
Wanneer de patiënt plaveiselcelcarcinoom van de huid (Cutaneous Squamous Cell Carcinoma, cuSCC) ontwikkelt, wordt het aanbevolen om de behandeling voort te zetten zonder aanpassing van de dosis vemurafenib (zie rubrieken 4.4 en 4.8).
Tabel 1: Schema voor doseringsaanpassing gebaseerd op de graad van iedere bijwerking (Adverse Event, AE)
Graad (CTC-AE)(a) | Aanbevolen doseringsaanpassing |
Graad 1 of graad 2 (draaglijk) | Handhaaf vemurafenib met een dosering van tweemaal daags 960 mg. |
Graad 2 (ondraaglijk) of graad 3 |
|
1e optreden van iedere graad 2 of 3 AE | Onderbreek behandeling tot verbetering naar graad 0 – 1. |
2e optreden van iedere graad 2 of 3 AE of persistentie na onderbreking van de behandeling | Onderbreek behandeling tot verbetering naar graad 0 – 1. |
3e optreden van iedere graad 2 of 3 AE of persistentie na een tweede dosisreductie | De behandeling definitief staken. |
Graad 4 |
|
1e optreden van iedere graad 4 AE | De behandeling definitief staken of onderbreek behandeling tot verbetering naar graad 0 – 1. |
2e optreden van iedere graad 4 AE of persistentie van iedere graad 4 AE na een eerste dosisreductie | De behandeling definitief staken. |
(a)De ernst van klinische bijwerkingen volgens ‘Common Terminology Criteria for Adverse Events’ v4.0 (CTC-AE) gradering.
Blootstellingsafhankelijke verlenging van het QT-interval werd gezien in een open-label fase II-studie zonder controlegroep bij patiënten met een gemetastaseerd melanoom die eerder behandeld werden. Voor het behandelen van QTc-verlenging kan het nodig zijn om specifieke controlemaatregelen te nemen (zie rubriek 4.4).
Tabel 2: Schema voor doseringsaanpassing gebaseerd op verlenging van het QT-interval
QTc-waarde | Aanbevolen doseringsaanpassing |
QTc> 500 ms bij baseline | Behandeling wordt niet aanbevolen |
Toename van QTc heeft een waarde van zowel > 500 ms als > 60 ms verandering van de waarde ten opzichte van voor de behandeling | De behandeling definitief staken |
1e optreden van QTc> 500 ms gedurende de behandeling en de verandering van de waarde ten opzichte van voor de behandeling blijft < 60 ms | Onderbreek behandeling tijdelijk totdat QTc lager is dan 500 ms. |
2e optreden van QTc> 500 ms gedurende de behandeling en de verandering van de waarde ten opzichte van voor de behandeling blijft < 60 ms | Onderbreek behandeling tijdelijk totdat QTc lager is dan 500 ms. |
3e optreden van QTc> 500 ms gedurende de behandeling en de verandering van de waarde ten opzichte van voor de behandeling blijft < 60 ms | De behandeling definitief staken. |
Speciale bevolkingsgroepen
Ouderen
Er is geen speciale doseringsaanpassing nodig voor patiënten > 65 jaar oud.
Patiënten met een verminderde nierfunctie
Er zijn beperkte gegevens beschikbaar bij patiënten met een verminderde nierfunctie. Een risico op toegenomen blootstelling bij patiënten met een ernstig verminderde nierfunctie kan niet worden uitgesloten. Patiënten met een ernstig verminderde nierfunctie moeten nauwlettend gecontroleerd worden (zie rubrieken 4.4 en 5.2).
Patiënten met een verminderde leverfunctie
Er zijn beperkte gegevens beschikbaar bij patiënten met een verminderde leverfunctie. Omdat vemurafenib door de lever geklaard wordt, kunnen patiënten met een matig tot ernstig verminderde leverfunctie een toegenomen blootstelling hebben en moeten ze nauwlettend gecontroleerd worden (zie rubrieken 4.4 en 5.2).
Pediatrische patiënten
De veiligheid en werkzaamheid van vemurafenib zijn niet vastgesteld bij kinderen onder de 18 jaar. De momenteel beschikbare gegevens worden beschreven in rubrieken 4.8, 5.1 en 5.2, maar er kan geen doseringsadvies worden gegeven.
Niet-blanke patiënten
De veiligheid en werkzaamheid van vemurafenib zijn niet vastgesteld bij niet-blanke patiënten. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Vemurafenib is voor oraal gebruik. De tabletten moeten in hun geheel met water doorgeslikt worden en mogen niet gekauwd of vermalen worden.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest voorkomende bijwerkingen (> 30%) van alle graden die bij vemurafenib werden gemeld zijn gewrichtspijn, vermoeidheid, uitslag, lichtgevoeligheidsreacties, alopecia, misselijkheid, diarree, hoofdpijn, pruritus, braken, papilloom van de huid en hyperkeratose. De meest voorkomende (≥ 5%) graad 3 bijwerkingen waren cuSCC, keratoacanthoom, uitslag, gewrichtspijn en verhoogd gamma-glutamyltransferase (GGT). CuSCC werd meestal behandeld door lokale excisie.
Tabel met een samenvatting van de bijwerkingen
Bijwerkingen die werden gemeld bij patiënten met melanoom zijn hieronder weergegeven naar systeem/orgaanklassen, frequentie en graad van ernst volgens MedDRA. De volgende termen zijn gebruikt voor de classificatie van frequentie:
Zeer vaak ≥ 1/10
Vaak ≥ 1/100, < 1/10
Soms ≥ 1/1.000, < 1/100
Zelden ≥ 1/10.000, < 1/1.000
Zeer zelden < 1/10.000
In deze rubriek zijn de bijwerkingen gebaseerd op resultaten van 468 patiënten uit een gerandomiseerde, open-label, fase III-studie bij volwassen patiënten met inoperabel of stadium IV-melanoom die positief zijn voor de BRAF V600-mutatie. Daarnaast zijn de bijwerkingen tevens gebaseerd op de resultaten van een fase II-studie met één behandelarm bij patiënten met stadium IV-melanoom die positief zijn voor de BRAF V600-mutatie, bij wie ten minste één voorafgaande systemische therapie gefaald had (zie rubriek 5.1). Verder worden er bijwerkingen gemeld die afkomstig zijn uit veiligheidsrapporten uit alle klinische studies en post-marketing gegevens. Alle opgenomen termen zijn gebaseerd op het hoogste percentage dat werd waargenomen in de fase II en fase III klinische studies. Binnen elke frequentiegroep worden bijwerkingen weergegeven in volgorde van afnemende ernst en deze werden gemeld met gebruik van de NCI-CTCAE v 4.0 (‘common toxicity criteria’) voor de beoordeling van toxiciteit.
Tabel 3: Bijwerkingen die optraden bij patiënten die behandeld werden met vemurafenib in de fase II- of fase III-studie en voorvallen afkomstig uit veiligheidsrapporten van alle studies(1) en gegevens na het in de handel brengen(2)
Systeem/orgaanklasse | Zeer vaak | Vaak | Soms | Zelden |
Infecties en parasitaire aandoeningen |
| Folliculitis |
|
|
Neoplasmata, benigne, maligne en niet-gespecificeerd (inclusief cysten en poliepen) | Plaveiselcelcarcinoom van de huid(d), keratoacanthoom, seborrhoïsche keratose, papilloom van de huid | Basaalcelcarcinoom, nieuw primair melanoom(3) | Niet-cuSCC(1)(3) | Chronische myelomonocytische leukemie(2)(4), pancreas adenocarcinoom(5) |
Bloed- en lymfestelselaandoeningen |
| Neutropenie, trombocytopenie(6) |
|
|
Immuunsysteemaandoeningen |
|
|
| Sarcoïdose(1)(2)(j) |
Voedings- en stofwisselingsstoornissen | Verminderde eetlust |
|
|
|
Zenuwstelselaandoeningen | Hoofdpijn, dysgeusie, duizeligheid | Paralyse 7e hersenzenuw, perifere neuropathie |
|
|
Oogaandoeningen |
| Uveïtis | Occlusie vena retina, iridocyclitis |
|
Bloedvataandoeningen |
| Vasculitis |
|
|
Ademhalings-stelsel-, borstkas- en mediastinumaandoeningen | Hoest |
|
|
|
Maagdarmstelselaandoeningen | Diarree, braken, misselijkheid, obstipatie | Stomatitis | Pancreatitis(2) |
|
Lever- en galaandoeningen |
|
| Leverschade(1)(2)(g) |
|
Huid- en onderhuidaandoeningen | Lichtgevoeligheidsreactie, actinische keratose, uitslag, maculopapulaire uitslag, pruritus, hyperkeratose, erytheem, palmoplantair erytrodysesthesiesyndroom, alopecia, droge huid, zonnebrand | Papulaire uitslag, panniculitis (inclusief erythema nodosum), keratosis pilaris | Toxische epidermale necrolyse(e), stevens-johnsonsyndroom(f) | Toxicodermie met eosinofilie en systemische symptomen(1)(2) |
Skeletspierstelsel- en bindweefselaandoeningen | Spierpijn, gewrichtspijn, pijn in de extremiteiten, skeletspierpijn, rugpijn | Artritis | Fibromatose van de fascia plantaris (1)(2), contractuur van Dupuytren(1)(2) |
|
Nier- en urinewegaandoeningen |
|
|
| Acute interstitiële nefritis(1)(2)(h), acute tubulaire necrose(1)(2)(h) |
Algemene aandoeningen en toedieningsplaatsstoornissen | Vermoeidheid, koorts, perifeer oedeem, asthenie |
|
|
|
Onderzoeken |
| Toename ALAT(c), toename alkalische fosfatase(c), toename ASAT(c), toename bilirubine(c), toename GGT(c), gewichtsverlies, QT-elektrocardiogram verlengd, toename creatinine in bloed(1)(2)(h) |
|
|
Letsels, intoxicaties en verrichtings-complicaties |
| Versterking van radiotoxiciteit (1)(2)(i) |
|
|
(1) Voorvallen afkomstig uit veiligheidsrapporten uit alle studies.
(2) Voorvallen afkomstig uit gegevens na het in de handel brengen.
(3) Een causaal verband tussen het geneesmiddel en de bijwerking is op zijn minst een aannemelijke mogelijkheid.
(4) Progressie van reeds bestaande NRAS-gemuteerde chronische myelomonocytische leukemie.
(5) Progressie van reeds bestaande KRAS-gemuteerde pancreas adenocarcinoom.
(6) Berekend op basis van fase II- en fase III-studies.
Omschrijving van specifieke bijwerkingen
Verhoging van leverenzymen(c)
Leverenzymafwijkingen die gemeld werden in de fase III klinische studie zijn hieronder weergegeven als het deel van de patiënten dat een verschuiving heeft ervaren van de leverenzymafwijkingen van baseline naar graad 3 of 4:
zeer vaak: GGT
vaak: ALAT, alkalische fosfatase, bilirubine
soms: ASAT
Er was geen verergering naar graad 4 ALAT, alkalische fosfatase of bilirubine.
Leverschade(g)
Op basis van de criteria voor leverschade veroorzaakt door geneesmiddelen, opgesteld door een internationale expert-werkgroep van clinici en wetenschappers, werd leverschade gedefinieerd als een van de volgende laboratoriumafwijkingen:
≥ 5 x ULN ALAT
≥ 2 x ULN alkalische fosfatase (zonder andere reden voor verhoogde alkalische fosfatase)
≥ 3 x ULN ALAT met gelijktijdig verhoogde concentratie bilirubine > 2 x ULN
Cutaan plaveiselcelcarcinoom(d) (cuSCC)
Er zijn gevallen van cuSCC gemeld bij patiënten die behandeld werden met vemurafenib. De incidentie van cuSCC was ongeveer 20% bij patiënten die met vemurafenib behandeld werden in studieverband. Het merendeel van de weggesneden laesies, beoordeeld door een onafhankelijk centraal dermatopathologisch laboratorium, werd geclassificeerd als SCC-keratoacanthomasubtype of met mixed-keratoacanthomakenmerken (52%). De meeste laesies die geclassificeerd werden als “overigen” (43%) waren goedaardige huidlaesies (bijv. verruca vulgaris, actinische keratose, benigne keratose, cyste/benigne cyste). CuSCC kwam meestal in een vroeg stadium van de behandeling voor met een mediane tijd tot de eerste verschijning van 7 tot 8 weken. Van de patiënten die cuSCC ontwikkelden, had ongeveer 33% > 1 incident met een mediane tijd van 6 weken tussen de incidenten. Gevallen van cuSCC werden over het algemeen behandeld met eenvoudige excisie, en patiënten vervolgden over het algemeen de behandeling zonder een doseringsaanpassing (zie rubrieken 4.2 en 4.4).
Niet-cutaan plaveiselcelcarcinoom (niet-cuSCC)
Er zijn meldingen ontvangen van niet-cuSCC bij patiënten die vemurafenib kregen terwijl zij waren opgenomen in een klinische studie. Controle op niet-cuSCC dient plaats te vinden zoals omschreven in rubriek 4.4.
Nieuw primair melanoom
In klinisch onderzoek zijn nieuwe primair maligne melanomen gerapporteerd. Deze melanomen werden weggesneden en de behandeling werd zonder doseringsaanpassing voortgezet. Controle op huidlaesies dient te gebeuren zoals beschreven in rubriek 4.4.
Versterking van radiotoxiciteit(i)
De meldingen omvatten "recall" fenomeen, huidschade door straling, stralingspneumonitis, stralingsoesofagitis, stralingsproctitis, stralingshepatitis, stralingscystitis en stralingsnecrose.
In een klinisch fase III-onderzoek (MO25515, n = 3219) werd een hogere incidentie van versterkte radiotoxiciteit gerapporteerd bij patiënten die voor en tijdens de behandeling met vemurafenib radiotherapie kregen (9,1%) vergeleken met patiënten die gelijktijdig vemurafenib en radiotherapie kregen (5,2%) of bij wie de radiotherapie voor de behandeling met vemurafenib werd gegeven (1,5%).
Overgevoeligheidsreacties(e)
Ernstige overgevoeligheidsreacties, waaronder anafylaxie, zijn gemeld in verband met vemurafenib. Ernstige overgevoeligheidsreacties zijn onder andere stevens-johnsonsyndroom, gegeneraliseerde uitslag, erytheem of hypotensie. Bij patiënten met ernstige overgevoeligheidsreacties dient de behandeling met vemurafenib definitief gestaakt te worden (zie rubriek 4.4).
Dermatologische reacties (f)
Ernstige dermatologische reacties, waaronder zeldzame gevallen van stevens-johnsonsyndroom en toxische epidermale necrolyse, zijn gerapporteerd bij patiënten die vemurafenib gebruikten in de klinische registratie-onderzoek. De behandeling met vemurafenib dient definitief gestaakt te worden bij patiënten die een ernstige dermatologische reactie ervaren.
Verlenging van het QT-interval
In een open-label fase II QT-substudie zonder controlegroep (NP22657) werd een analyse uitgevoerd van gecentraliseerde ECG-gegevens van 132 patiënten die gedoseerd werden met tweemaal daags 960 mg vemurafenib. Deze analyse toonde een blootstellingsafhankelijke verlenging van het QTc-interval. Het gemiddelde QTc-effect bleef stabiel tussen 12-15 ms tot voorbij de eerste behandelmaand, waarbij de grootste gemiddelde verlenging van het QTc-interval (15,1 ms; bovengrens 95%-BI: 17,7 ms) werd waargenomen binnen de eerste 6 maanden (n = 90 patiënten). Twee patiënten (1,5%) ontwikkelden absolute QTc-waarden van > 500 ms (CTC-graad 3) opkomend tijdens behandeling. Slechts één patiënt (0,8%) vertoonde een QTc-verandering van > 60 ms vanaf baseline (zie rubriek 4.4).
Acute nierschade(h)
Gevallen van niertoxiciteit zijn gemeld bij vemurafenib, variërend van verhoogd creatinine tot acute interstitiële nefritis en acute tubulaire necrose. Enkele daarvan werden samen met dehydratie gemeld. Verhoogd serumcreatinine was meestal licht (>1‑1,5 x ULN) tot matig (>1,5‑3 x ULN) van aard en reversibel (zie tabel 4).
Tabel 4: Verandering in creatinine vanaf baseline in het fase III-onderzoek
| Vemurafenib (%) | Dacarbazine (%) |
Verandering 1 graad vanaf baseline tot elke graad | 27,9 | 6,1 |
Verandering 1 graad vanaf baseline tot graad 3 of hoger | 1,2 | 1,1 |
Tot graad 3 | 0,3 | 0,4 |
Tot graad 4 | 0,9 | 0,8 |
Tabel 5: Gevallen van acute nierschade in het fase III-onderzoek
| Vemurafenib (%) | Dacarbazine (%) |
Gevallen van acute nierschade* | 10,0 | 1,4 |
Gevallen van acute nierschade samen met dehydratie gemeld | 5,5 | 1,0 |
Doseringsaanpassing wegens acute nierschade | 2,1 | 0 |
Alle percentages worden weergegeven als gevallen van het totaal aantal patiënten dat is blootgesteld aan elk geneesmiddel.
*Omvat acute nierschade, verminderde nierfunctie en veranderde laboratoriumwaarden overeenkomend met acute nierschade.
Sarcoïdose(j)
Gevallen van sarcoïdose, waarbij voornamelijk de huid, longen en ogen getroffen waren, zijn gemeld bij patiënten behandeld met vemurafenib. In de meeste gevallen werd doorbehandeld met vemurafenib en de sarcoïdose verdween of hield aan.
Speciale bevolkingsgroepen
Ouderen
In de fase III-studie waren 94 (28%) van de 336 patiënten met inoperabel of gemetastaseerd melanoom die behandeld werden met vemurafenib ≥ 65 jaar. Oudere patiënten (≥ 65 jaar) zijn mogelijk gevoeliger voor bijwerkingen, waaronder cuSCC, verminderde eetlust en hartaandoeningen.
Geslacht
Graad 3-bijwerkingen die tijdens de klinische studies met vemurafenib vaker werden gemeld bij vrouwen dan mannen waren uitslag, gewrichtspijn en lichtgevoeligheid.
Pediatrische patiënten
De veiligheid van vemurafenib is niet vastgesteld bij kinderen en adolescenten. In een klinisch onderzoek met 6 adolescente patiënten werden geen nieuwe veiligheidssignalen waargenomen.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden (zie hieronder voor details).
België
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Duitsland
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/12/751/001
10. DATUM VAN HERZIENING VAN DE TEKST
18 maart 2024
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau (http://www.ema.europa.eu).
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 2910354 | ZELBORAF 240 MG FILMOMH TABL 56 X 240 MG UD | L01EC01 | - | € 1353,13 | Ja | - | - |