SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
Volibris 2,5 mg filmomhulde tabletten
Volibris 5 mg filmomhulde tabletten
Volibris 10 mg filmomhulde tabletten
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Volibris 2,5 mg filmomhulde tabletten
Elke tablet bevat 2,5 mg ambrisentan.
Hulpstof(fen) met bekend effect
Elke tablet bevat ongeveer 92,6 mg lactose (als monohydraat) en ongeveer 0,25 mg lecithine (soja) (E322).
Volibris 5 mg filmomhulde tabletten
Elke tablet bevat 5 mg ambrisentan.
Hulpstof(fen) met bekend effect
Elke tablet bevat ongeveer 90,3 mg lactose (als monohydraat), ongeveer 0,25 mg lecithine (soja) (E322) en ongeveer 0,11 mg allurarood AC aluminiumlak (E129).
Volibris 10 mg filmomhulde tabletten
Elke tablet bevat 10 mg ambrisentan.
Hulpstof(fen) met bekend effect:
Elke tablet bevat ongeveer 85,5 mg lactose (als monohydraat), ongeveer 0,25 mg lecithine (soja) (E322) en ongeveer 0,45 mg allurarood AC aluminiumlak (E129).
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Filmomhulde tablet (tablet).
Volibris 2,5 mg filmomhulde tabletten
Witte, ronde, convexe, filmomhulde tablet van 7 mm met aan de ene zijde de markering “GS” en aan de andere zijde “K11”.
Volibris 5 mg filmomhulde tabletten
Lichtroze, vierkante, convexe, filmomhulde tablet van 6,6 mm met aan de ene zijde de markering “GS” en aan de andere zijde “K2C”.
Volibris 10 mg filmomhulde tabletten
Dieproze, ovale, convexe, filmomhulde tablet van 9,8 × 4.9 mm met aan de ene zijde de markering “GS” en aan de andere zijde “KE3”.
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Volibris is geïndiceerd voor de behandeling van pulmonale arteriële hypertensie (PAH) bij volwassen patiënten geclassificeerd als WHO functionele klasse (FC) II of III, waaronder gebruik in een combinatietherapie (zie rubriek 5.1). De werkzaamheid is aangetoond bij idiopathische PAH (IPAH) en bij PAH geassocieerd met bindweefselaandoening.
Volibris is geïndiceerd voor de behandeling van PAH bij adolescenten en kinderen (van 8 tot jonger dan 18 jaar) geclassificeerd als WHO functionele klasse (FC) II of III, waaronder gebruik in een combinatietherapie. De werkzaamheid is aangetoond bij IPAH, bij familiaire, gecorrigeerde congenitale PAH en bij PAH geassocieerd met bindweefselaandoening (zie rubriek 5.1).
4.2 Dosering en wijze van toediening
De behandeling dient ingesteld te worden door een arts die ervaring heeft met de behandeling van PAH.
Dosering
Volwassenen
Ambrisentan als monotherapie
Volibris dient oraal te worden ingenomen in een startdosering van 5 mg eenmaal daags. Deze startdosering mag worden verhoogd tot 10 mg eenmaal daags, op basis van de klinische respons en de verdraagbaarheid.
Ambrisentan in combinatie met tadalafil
Indien het in combinatie met tadalafil wordt gebruikt, moet Volibris getitreerd worden tot 10 mg eenmaal daags.
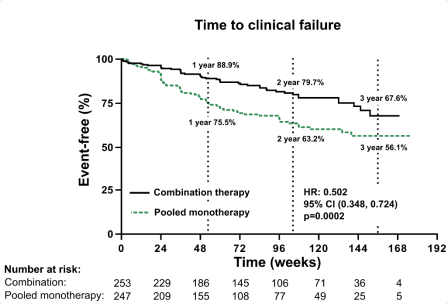
In het AMBITION-onderzoek kregen patiënten gedurende de eerste acht weken 5 mg ambrisentan per dag, waarna dit werd getitreerd tot 10 mg, op basis van verdraagbaarheid (zie rubriek 5.1). Indien het in combinatie met tadalafil werd gebruikt, kregen patiënten als startdosering 5 mg ambrisentan en 20 mg tadalafil. Op basis van de verdraagbaarheid werd de dosis tadalafil na 4 weken getitreerd naar 40 mg en werd de dosis ambrisentan na 8 weken getitreerd naar 10 mg. Dit werd bij meer dan 90% van de patiënten bereikt. De doses konden ook worden verlaagd, op basis van verdraagbaarheid.
Er zijn beperkte gegevens die erop duiden dat plotseling stoppen van ambrisentan niet geassocieerd kan worden met een rebound verergering van PAH.
Ambrisentan in combinatie met ciclosporine A
Indien ambrisentan bij volwassenen wordt toegediend in combinatie met ciclosporine A moet de dosering worden beperkt tot 5 mg eenmaal daags en moet de patiënt goed gecontroleerd worden (zie rubrieken 4.5 en 5.2).
Pediatrische patiënten van 8 tot en met 17 jaar
Ambrisentan als monotherapie of in combinatie met andere therapieën tegen PAH
Volibris dient oraal te worden ingenomen op basis van het hieronder beschreven doseerschema:
Lichaamsgewicht (kg) | Eenmaaldaagse startdosis (mg) | Daaropvolgende eenmaaldaagse dosistitratie (mg)a |
≥ 50 | 5 | 10 |
≥ 35 tot < 50 | 5 | 7,5 |
≥ 20 tot < 35 | 2,5 | 5 |
a = op basis van klinische respons en verdraagbaarheid (zie rubriek 5.1) | ||
Ambrisentan in combinatie met ciclosporine A
Indien ambrisentan bij pediatrische patiënten wordt toegediend in combinatie met ciclosporine A moet de dosering worden beperkt tot 5 mg eenmaal daags voor patiënten van ≥ 50 kg, of tot 2,5 mg eenmaal daags voor patiënten van ≥ 20 tot < 50 kg. De patiënt moet goed gecontroleerd worden (zie rubrieken 4.5 en 5.2).
Bijzondere populaties
Oudere patiënten
Er is geen dosisaanpassing nodig voor patiënten vanaf 65 jaar (zie rubriek 5.2).
Patiënten met een verminderde nierfunctie
Er is geen dosisaanpassing nodig voor patiënten met een verminderde nierfunctie (zie rubriek 5.2). Er is beperkte ervaring met ambrisentan bij personen met een ernstig verminderde nierfunctie (creatinineklaring < 30 ml/min); bij deze subgroep dient de behandeling met de nodige voorzichtigheid gestart te worden en dient men bijzonder voorzichtig te zijn als de dosering wordt verhoogd tot 10 mg ambrisentan.
Patiënten met een verminderde leverfunctie
Ambrisentan is niet onderzocht bij personen met een afgenomen leverfunctie (met of zonder cirrose). De belangrijkste klaringsroutes van ambrisentan zijn glucuronidatie en oxidatie met de daarop volgende eliminatie in de gal. Daarom kan bij een verminderde leverfunctie verwacht worden, dat de blootstelling (Cmax en AUC) aan ambrisentan verhoogd zou kunnen zijn. Vandaar dat niet met ambrisentan gestart moet worden bij patiënten met een ernstig verminderde leverfunctie, of met klinisch significant verhoogde leveraminotransferasen (hoger dan driemaal de normaalwaarde bovengrens (>3xBGN); zie rubrieken 4.3 en 4.4).
Pediatrische patiënten
De veiligheid en werkzaamheid van ambrisentan bij kinderen jonger dan 8 jaar zijn niet vastgesteld. Er zijn geen klinische gegevens beschikbaar (zie rubriek 5.3 voor beschikbare gegevens in jonge dieren).
Wijze van toediening
Volibris is voor oraal gebruik. Het wordt aanbevolen de tablet in zijn geheel door te slikken; de tablet kan met of zonder voedsel ingenomen worden. Het wordt aanbevolen de tablet niet te breken, vermalen of erop te kauwen.
4.3 Contra-indicaties
• Overgevoeligheid voor het werkzame bestanddeel, voor soja, of voor één van de in rubriek 6.1 vermelde hulpstoffen.
• Zwangerschap (zie rubriek 4.6)
• Vrouwen in de vruchtbare leeftijd die geen betrouwbare anticonceptiemethode gebruiken (zie rubrieken 4.4 en 4.6).
• Borstvoeding (zie rubriek 4.6)
• Ernstig verminderde leverfunctie (met of zonder cirrose) (zie rubriek 4.2)
• Uitgangswaarden van leveraminotransferasen (aspartaat aminotransferasen (AST) en/of alanine aminotransferasen (ALT)) > 3xBGN (zie rubrieken 4.2 en 4.4)
• Idiopathische pulmonale fibrose (IPF), met of zonder secundaire pulmonale hypertensie (zie rubriek 5.1)
4.8 Bijwerkingen
Samenvatting van het bijwerkingenprofiel
Perifeer oedeem (37%) en hoofdpijn (28%) waren de meest vaak voorkomende bijwerkingen die met ambrisentan zijn waargenomen. De hogere dosis (10 mg) werd in verband gebracht met een hogere incidentie van deze bijwerkingen, en perifeer oedeem neigt bij patiënten van 65 jaar en ouder ernstiger te zijn in kortdurende klinische onderzoeken (zie rubriek 4.4).
Ernstige bijwerkingen geassocieerd met het gebruik van ambrisentan omvatten anemie (verlaagde hemoglobinewaarde, verlaagde hematocrietwaarde) en hepatotoxiciteit.
Afnames in de hemoglobine- en hematocrietconcentraties (10%) zijn geassocieerd met ERA’s, inclusief ambrisentan. De meeste van deze afnames werden waargenomen tijdens de eerste vier weken van behandeling en over het algemeen stabiliseerde de hemoglobine daarna (zie rubriek 4.4).
Verhoging van leverenzymen (2%), leverschade en auto-immuunhepatitis (waaronder exacerbatie van onderliggende ziekte) zijn waargenomen met ambrisentan (zie rubrieken 4.4 en 5.1).
Tabel met bijwerkingen
De frequentiegroepen zijn gedefinieerd als: zeer vaak (≥ 1/10), vaak (≥ 1/100 tot < 1/10), soms (≥ 1/1.000 tot < 1/100), zelden (≥ 1/10.000 tot < 1/1.000), zeer zelden (< 1/10.000) en niet bekend (kan met de beschikbare gegevens niet worden bepaald). Voor dosisgerelateerde bijwerkingen geeft de frequentiegroep de hogere dosering van ambrisentan weer. Binnen elke frequentiecategorie worden de bijwerkingen weergegeven naar afnemende ernst.
Systeem/orgaanklasse | Frequentie | Bijwerking(en) |
Bloed‑ en lymfestelselaandoeningen | Zeer vaak | Anemie (verlaagde hemoglobinewaarde, |
Immuunsysteemaandoeningen | Vaak | Overgevoeligheidsreacties (bijv. angio-oedeem, rash, pruritus) |
Zenuwstelselaandoeningen | Zeer vaak | Hoofdpijn (waaronder sinushoofdpijn, migraine)2, |
Oogaandoeningen | Vaak | Wazig zien, |
Evenwichtsorgaan‑ en ooraandoeningen | Vaak | Tinnitus3 |
Soms | Plotseling gehoorverlies3 | |
Hartaandoeningen | Zeer vaak | Palpitaties |
Vaak | Hartfalen4 | |
Bloedvataandoeningen | Zeer vaak | Blozen5 |
Vaak | Hypotensie, | |
Ademhalingsstelsel‑, borstkas‑ en mediastinumaandoeningen | Zeer vaak | Dyspneu6, |
Vaak | Epistaxis, | |
Maagdarmstelselaandoeningen | Zeer vaak | Misselijkheid, |
Vaak | Buikpijn, | |
Lever‑ en galaandoeningen | Vaak | Levertransaminasen verhoogd |
Soms | Leverschade (zie rubriek 4.4), | |
Huid‑ en onderhuidaandoeningen | Vaak | Rash8 |
Algemene aandoeningen en toedieningsplaatsstoornissen | Zeer vaak | Perifeer oedeem, |
Vaak | Asthenie |
1 Zie rubriek Omschrijving van geselecteerde bijwerkingen
2 De frequentie van hoofdpijn leek hoger met 10 mg ambrisentan.
3 Gevallen werden alleen waargenomen in een placebogecontroleerd klinisch onderzoek naar ambrisentan in combinatie met tadalafil.
4 De meeste gevallen waarin hartfalen werd gemeld gingen gepaard met vochtretentie.
5 De frequenties werden gezien in een placebogecontroleerd klinisch onderzoek naar ambrisentan in combinatie met tadalafil. Een lagere incidentie werd gezien bij ambrisentan als monotherapie.
6 Gevallen van verergering van dyspneu met een onduidelijke oorsprong zijn gemeld kort na het starten van de behandeling met ambrisentan.
7 Het optreden van nasale verstopping was dosisgerelateerd tijdens de behandeling met ambrisentan.
8 Rash omvat erythemateuze rash, gegeneraliseerde rash, papulaire rash en pruritische rash.
Omschrijving van geselecteerde bijwerkingen
Verlaagde hemoglobine
In de postmarketing periode zijn gevallen van anemie gemeld waarvoor een bloedtransfusie nodig was (zie rubriek 4.4). De frequentie van verlaagde hemoglobine (anemie) was hoger met 10 mg ambrisentan. Tijdens de 12 weken durende placebo-gecontroleerde fase 3 klinische onderzoeken namen de gemiddelde hemoglobineconcentraties bij patiënten in de ambrisentan-groepen af; deze werden al in week 4 waargenomen (afgenomen met 0,83 g/dl). De gemiddelde veranderingen ten opzichte van de beginwaarden leken zich te stabiliseren tijdens de volgende acht weken. In totaal kwamen bij 17 patiënten (6,5%) in de ambrisentan behandelgroepen verlagingen van de hemoglobine voor van ≥15% ten opzichte van de beginwaarden, en die onder de normaalwaarde ondergrens vielen.
Pediatrische patiënten
De veiligheid van ambrisentan bij pediatrische patiënten met PAH van 8 tot en met 17 jaar werd beoordeeld bij 41 patiënten die werden behandeld met ambrisentan 2,5 mg of 5 mg eenmaal daags (groep met lage dosering) of met ambrisentan 2,5 mg of 5 mg eenmaal daags getitreerd tot 5 mg, 7,5 mg of 10 mg op basis van lichaamsgewicht (groep met hoge dosering) alleen of in combinatie met andere geneesmiddelen voor PAH gedurende 24 weken in een open-label fase 2b-onderzoek. De veiligheid werd verder beoordeeld in een langetermijn-extensieonderzoek bij 38 van de 41 personen. De waargenomen bijwerkingen die werden beoordeeld als gerelateerd aan ambrisentan kwamen overeen met de bijwerkingen die zijn gezien in gecontroleerde onderzoeken met volwassen patiënten, waarbij hoofdpijn (15%, 6/41 personen gedurende de 24 weken van het open-label fase 2b-onderzoek en 8%, 3/38 personen gedurende het langetermijn-extensieonderzoek) en neusverstopping (7%, 3/41 personen gedurende de 24 weken van het open-label fase 2b-onderzoek) het vaakst optraden.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden
via het nationale meldsysteem:
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
www.fagg.be
Afdeling Vigilantie:
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg-afmps.be
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
GlaxoSmithKline Trading Services Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Ierland
D24 YK11
8. NUMMERS VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Volibris 2,5 mg filmomhulde tabletten
EU/1/08/451/005
Volibris 5 mg filmomhulde tabletten
EU/1/08/451/001
EU/1/08/451/002
Volibris 10 mg filmomhulde tabletten
EU/1/08/451/003
EU/1/08/451/004
10. DATUM VAN HERZIENING VAN DE TEKST
16/04/2026 (v36).
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig | Remgeld reguliere tegemoetkoming | Remgeld verhoogde tegemoetkoming |
|---|---|---|---|---|---|---|---|
| 2501278 | VOLIBRIS 10 MG COMP 30 X 10 MG | C02KX02 | - | € 903,83 | Ja | - | - |
| 2501286 | VOLIBRIS 5 MG COMP 30 X 5 MG | C02KX02 | - | € 903,83 | Ja | - | - |