ANNEXE I
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Que contient cette notice ?
- 1. DÉNOMINATION DU MÉDICAMENT
- 2. COMPOSITION QUALITATIVE ET QUANTITATIVE
- 3. FORME PHARMACEUTIQUE
- 4. INFORMATIONS CLINIQUES
- 4.1. Indications thérapeutiques
- 4.2. Posologie et mode d’administration
- 4.3. Contre-indications
- 4.4. Mises en garde spéciales et précautions d’emploi
- 4.5. Interactions avec d’autres médicaments et autres formes d’interactions
- 4.6. Fertilité, grossesse et allaitement
- 4.7. Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
- 4.8. Effets indésirables
- 4.9. Surdosage
- 5. PROPRIÉTÉS PHARMACOLOGIQUES
- 5.1. Propriétés pharmacodynamiques
- 5.2. Propriétés pharmacocinétiques
- 5.3. Données de sécurité préclinique
- 6. DONNÉES PHARMACEUTIQUES
- 6.1. Liste des excipients
- 6.2. Incompatibilités
- 6.3. Durée de conservation
- 6.4. Précautions particulières de conservation
- 6.5. Nature et contenu de l’emballage extérieur
- 6.6. Précautions particulières d’élimination et manipulation
- 7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
- 8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
- 9. DATE DE PREMIÈRE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
- 10. DATE DE MISE A JOUR DU TEXTE
- 10. PRÉCAUTIONS PARTICULIÈRES D’ÉLIMINATION DES MÉDICAMENTS NON UTILISÉS OU DES DÉCHETS PROVENANT DE CES MÉDICAMENTS S’IL Y A LIEU
- 11. NOM ET ADRESSE DU TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
- 12. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
- 13. NUMÉRO DU LOT
- 14. CONDITIONS DE PRESCRIPTION ET DE DÉLIVRANCE
- 15. INDICATIONS D’UTILISATION
- 16. INFORMATIONS EN BRAILLE
- 17. IDENTIFIANT UNIQUE - CODE-BARRES 2D
- 18. IDENTIFIANT UNIQUE - DONNÉES LISIBLES PAR LES HUMAINS
1. DÉNOMINATION DU MÉDICAMENT
YAXWER 120 mg solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 120 mg de denosumab dans au moins 1,7 mL de solution (70 mg/mL).
Le denosumab est un anticorps monoclonal IgG2 humain produit dans une lignée cellulaire de mammifère (cellules d’ovaires de hamster chinois) par la technique de l’ADN recombinant.
Excipient à effet notoire :
Chaque 1,7 mL de solution contient 78 mg de sorbitol (E420) et 0,17 mg de polysorbate 20 (E 432).
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable.
Solution limpide, incolore à légèrement jaune, exempte de particules visibles.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Prévention des complications osseuses (fractures pathologiques, irradiation osseuse, compression médullaire ou chirurgie osseuse) chez des patients adultes présentant une affection maligne avancée avec atteinte osseuse (voir rubrique 5.1).
Traitement des adultes et des adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes, non résécables ou pour lesquels la résection chirurgicale est susceptible d’entraîner une morbidité sévère.
4.2 Posologie et mode d’administration
YAXWER doit être administré sous la responsabilité d’un professionnel de santé. Posologie
Une supplémentation quotidienne apportant au moins 500 mg de calcium et 400 UI de vitamine D est requise chez tous les patients, sauf en cas d’hypercalcémie (voir rubrique 4.4).
Les patients traités par YAXWER devront recevoir la notice et la carte d’information au patient.
Prévention des complications osseuses chez les patients adultes présentant une affection maligne avancée avec atteinte osseuse
La posologie recommandée est de 120 mg, administrée une fois toutes les quatre semaines, par injection sous-cutanée dans la cuisse, l’abdomen ou le bras.
Tumeur osseuse à cellules géantes
La posologie recommandée est de 120 mg de YAXWER toutes les quatre semaines administrées en une seule injection par voie sous-cutanée dans la cuisse, l’abdomen ou le bras avec une dose supplémentaire de 120 mg aux jours 8 et 15 du premier mois de traitement.
Au cours d’un essai de phase II les patients qui ont subi une résection complète de la tumeur osseuse à cellules géantes ont reçu le traitement pendant 6 mois supplémentaires après la chirurgie conformément au protocole.
Les patients atteints de tumeurs osseuses à cellules géantes doivent être examinés à intervalles réguliers pour déterminer si le traitement leur est toujours bénéfique. Chez les patients dont la maladie est contrôlée par YAXWER, l’effet de l’interruption ou de l’arrêt du traitement n’a pas été évalué, cependant des données limitées chez ces patients n’indiquent pas de rechute de la maladie à l’arrêt du traitement.
Insuffisance rénale
Aucune adaptation de la posologie n’est nécessaire chez les patients atteints d’insuffisance rénale (voir les rubriques 4.4 pour les recommandations relatives à la surveillance de la calcémie, et les sections 4.8 et 5.2).
Insuffisance hépatique
La sécurité et l’efficacité du denosumab n’ont pas été étudiées chez les patients présentant une insuffisance hépatique (voir rubrique 5.2).
Patients âgés (âge ≥ 65 ans)
Aucune adaptation de la posologie n’est nécessaire chez les patients âgés (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de YAXWER n’ont pas été établies dans la population pédiatrique (âge
< 18 ans), excepté chez les adolescents (âgés de 12 à 17 ans) à maturité squelettique atteints de tumeurs osseuses à cellules géantes.
YAXWER n’est pas recommandé chez l’enfant (âge < 18 ans), excepté chez les adolescents (âgés de 12 à 17 ans) à maturité squelettique atteints de tumeurs osseuses à cellules géantes (voir rubrique 4.4).
Traitement des adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes non résécables ou pour lesquels la résection chirurgicale est susceptible d’entraîner une morbidité sévère : la posologie est la même que chez l’adulte.
Chez l’animal, l’inhibition de RANK/RANK-ligand (RANKL) a été associée à une inhibition de la croissance osseuse et à une absence de poussée dentaire. Ces modifications ont été partiellement réversibles à l’arrêt de l’inhibition de RANKL (voir rubrique 5.3).
Mode d’administration
Injection sous-cutanée.
YAXWER 120 mg/1,7 mL solution en flacon à usage unique :
L’administration du flacon de 120 mg/1,7 mL ne doit être effectuée que par un professionnel de santé.
Pour les instructions concernant l’utilisation, la manipulation et l’élimination, voir rubrique 6.6.
4.3 Contre-indications
- Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
- Hypocalcémie sévère non traitée (voir rubrique 4.4).
- Lésions non cicatrisées résultant d’une chirurgie bucco-dentaire.
4.4 Mises en garde spéciales et précautions d’emploi
Traçabilité
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro du lot du produit administré doivent être clairement enregistrés.
Supplémentation en calcium et vitamine D
Une supplémentation en calcium et en vitamine D est requise chez tous les patients, sauf en cas d’hypercalcémie (voir rubrique 4.2).
Hypocalcémie
Avant l’instauration du traitement par YAXWER, toute hypocalcémie préexistante doit être corrigée. Une hypocalcémie peut survenir à tout moment pendant le traitement par YAXWER. La surveillance de la calcémie doit être effectuée (i) avant la première injection de YAXWER, (ii) deux semaines après la première injection, (iii) si des symptômes d’hypocalcémie surviennent (voir rubrique 4.8 pour les symptômes). Une surveillance supplémentaire de la calcémie doit être envisagée pendant le traitement chez les patients ayant des facteurs de risque d’hypocalcémie, ou selon l’état clinique du patient.
Les patients doivent être informés sur la nécessité de signaler tout symptôme pouvant faire suspecter une hypocalcémie. Une supplémentation additionnelle en calcium et une surveillance supplémentaire peuvent être nécessaires si une hypocalcémie survient au cours d’un traitement par YAXWER.
Des cas d’hypocalcémie symptomatique sévère (incluant des cas d’issue fatale) ont été rapportés après la commercialisation de YAXWER (voir rubrique 4.8), la plupart des cas se sont produits dans les premières semaines du traitement, mais ils peuvent survenir plus tard.
Insuffisance rénale
Les patients insuffisants rénaux sévères (clairance de la créatinine < 30 mL/min) ou dialysés présentent un risque accru d’hypocalcémie. Le risque de développer une hypocalcémie et une élévation conjointe de l’hormone parathyroïdienne augmentent avec le degré d’insuffisance rénale. Une surveillance régulière du taux de calcium est particulièrement importante chez ces patients.
Ostéonécrose de la mâchoire (ONM)
Une ONM a fréquemment été rapportée chez les patients traités par le denosumab (voir rubrique 4.8).
L’instauration d’un traitement/d’une nouvelle phase de traitement doit être retardée chez les patients présentant des lésions des tissus mous non cicatrisées dans la bouche. Un examen dentaire avec des soins préventifs et une évaluation individuelle du bénéfice/risque sont recommandés avant d’initier un traitement par le denosumab.
Lors de l’évaluation du risque de développer une ONM chez le patient, les facteurs de risque suivants doivent être considérés :
- puissance de la thérapie inhibitrice de la résorption osseuse (risque plus élevé lorsque le composé est puissant), voie d’administration (risque plus élevé lors d’une administration parentérale) et dose cumulée de traitement anti-résorptif osseux.
- cancer, présence de comorbidités (par exemple anémie, coagulopathies, infections), tabagisme.
- traitements concomitants : corticoïdes, chimiothérapies, inhibiteurs de l’angiogenèse, radiothérapie de la tête et du cou.
- mauvaise hygiène bucco-dentaire, affection parodontale, prothèse dentaire mal ajustée, affection dentaire préexistante, intervention dentaire invasive (par exemple extraction dentaire).
Tous les patients doivent être encouragés à maintenir une bonne hygiène bucco-dentaire, à faire des bilans dentaires réguliers et à signaler immédiatement tout symptôme oral tel que mobilité dentaire, douleur ou gonflement, ulcères non-cicatrisés ou écoulement au cours du traitement par le denosumab. Pendant le traitement, les interventions dentaires invasives ne doivent être effectuées qu’après un examen approfondi et doivent être évitées à proximité d’une administration d’une dose de denosumab.
La prise en charge des patients qui développent une ONM doit être mise en place en collaboration étroite entre le médecin traitant et le dentiste ou le chirurgien-dentiste ayant une expertise dans l’ONM. Lorsque cela est possible, une interruption temporaire du traitement par YAXWER doit être envisagée jusqu’à guérison complète de l’ONM et à la réduction des facteurs de risque.
Ostéonécrose du conduit auditif externe
L’ostéonécrose du conduit auditif externe a été rapportée avec le denosumab. Les facteurs de risque éventuels d’ostéonécrose du conduit auditif externe comprennent l’utilisation de stéroïdes et la chimiothérapie et/ou les facteurs de risque locaux tels qu’une infection ou un traumatisme. La possibilité d’ostéonécrose du conduit auditif externe doit être envisagée chez les patients recevant du denosumab qui présentent des symptômes auditifs, notamment des infections chroniques de l’oreille.
Fractures atypiques du fémur
Des fractures fémorales atypiques ont été rapportées chez des patients traités par le denosumab (voir rubrique 4.8). Les fractures fémorales atypiques sont des fractures des régions sous-trochantériennes et diaphysaires du fémur pouvant survenir suite à un traumatisme minime ou même sans traumatisme.
Ces fractures sont caractérisées par des aspects radiologiques spécifiques. Des fractures fémorales atypiques ont aussi été observées chez des patients présentant certaines comorbidités (par exemple carence en vitamine D, polyarthrite rhumatoïde, hypophosphatasie) et chez des patients traités par certains médicaments (par exemple des bisphosphonates, des glucocorticoïdes, des inhibiteurs de la pompe à protons). Ces évènements sont également survenus sans traitement inhibiteur de la résorption osseuse. Les fractures similaires observées lors de traitement par les bisphosphonates sont souvent bilatérales ; par conséquent, le fémur controlatéral doit être examiné chez les patients traités par le denosumab ayant eu une fracture fémorale diaphysaire. L’arrêt du traitement de denosumab chez les patients chez lesquels une fracture fémorale atypique est suspectée, doit être envisagé en fonction de l’évaluation du rapport bénéfice/risque pour le patient. Pendant le traitement par le denosumab, les patients doivent être informés que toute douleur nouvelle ou inhabituelle au niveau de la cuisse, de la hanche ou de l’aine doit être rapportée. Les patients présentant de tels symptômes devront être examinés pour rechercher une fracture fémorale atypique incomplète.
Hypercalcémie après l’arrêt du traitement chez les patients atteints de tumeurs osseuses à cellules géantes et chez les patients dont le squelette est en croissance
Des hypercalcémies cliniquement significatives nécessitant une hospitalisation et compliquées par une lésion rénale aiguë ont été rapportées quelques semaines à quelques mois après l’arrêt du traitement chez des patients atteints de tumeurs osseuses à cellules géantes traités par denosumab.
Après l’arrêt du traitement, surveiller les signes et symptômes d’une hypercalcémie chez les patients, envisager une évaluation régulière du calcium sérique, et réévaluer les besoins de supplémentation en calcium et en vitamine D du patient (voir rubrique 4.8).
YAXWER n’est pas recommandé chez les patients dont le squelette est en croissance (voir rubrique 4.2). Des hypercalcémies cliniquement significatives ont également été rapportées, quelques semaines à quelques mois après l’arrêt du traitement, dans ce groupe de patients.
Autres
Les patients recevant YAXWER ne doivent pas être traités simultanément avec d’autres médicaments contenant du denosumab (indiqués dans l’ostéoporose).
Les patients traités par YAXWER ne doivent pas être traités simultanément avec des bisphosphonates.
Des tumeurs osseuses à cellules géantes malignes ou la progression vers un état métastatique sont un évènement peu fréquent et un risque connu chez les patients atteints de tumeurs osseuses à cellules géantes. Les signes radiologiques de malignité, une nouvelle radiotransparence ou ostéolyse doivent être surveillés chez ces patients. Les données cliniques disponibles ne suggèrent pas de risque accru de malignité chez les patients atteints de tumeurs osseuses à cellules géantes traités par denosumab.
Excipients
Ce médicament contient 78 mg de sorbitol dans chaque dose (1,7 mL). L’effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l’apport alimentaire de sorbitol (ou de fructose) doit être pris en compte.
Ce médicament contient 0,17 mg de polysorbate 20 dans chaque dose (1,7 ml). Les polysorbates peuvent provoquer des réactions allergiques. Informez votre médecin si vous avez ou si votre enfant a des allergies connues.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose (1,7 mL), c.-à-d. qu’il est essentiellement « sans sodium ».
4.5 Interactions avec d’autres médicaments et autres formes d’interactions
Aucune étude d’interaction n’a été réalisée.
Lors des essais cliniques, le denosumab a été administré en association avec un traitement anticancéreux standard et chez des patients précédemment traités par bisphosphonates. Les paramètres pharmacocinétiques et pharmacodynamiques du denosumab (N-telopeptide urinaire corrigé par la créatinine, uNTx/Cr) n’ont pas été altérés par une chimiothérapie et/ou une hormonothérapie concomitante, ni par une précédente exposition à un bisphosphonate intraveineux.
4.6 Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation du denosumab chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3).
YAXWER n’est pas recommandé chez la femme enceinte ni chez la femme en âge de procréer et n’utilisant pas de contraception. Il doit être conseillé aux femmes de ne pas débuter une grossesse pendant le traitement par YAXWER et durant au moins 5 mois après le traitement. Les effets du denosumab sont plus susceptibles d’apparaître au cours du deuxième et du troisième trimestre de la grossesse car les anticorps monoclonaux circulent à travers le placenta de façon linéaire comparativement à l’avancée de la grossesse, avec la plus grande quantité transférée au cours du troisième trimestre.
Allaitement
On ne sait pas si le denosumab est excrété dans le lait maternel. Un risque pour les
nouveau-nés/nourrissons ne peut être exclu. Les études réalisées chez des souris knockout suggèrent que l'absence de RANKL au cours de la gestation peut perturber la maturation de glandes mammaires, entraînant une altération de l’allaitement post-partum (voir rubrique 5.3). Une décision doit être prise, soit de renoncer à allaiter, soit de renoncer à recevoir un traitement par YAXWER en prenant en compte le bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Aucune donnée n’est disponible concernant l’effet du denosumab sur la fertilité humaine. Les études chez l’animal n’ont pas montré d'effets délétères directs ou indirects sur la fertilité (voir rubrique 5.3.).
4.7 Effets sur l’aptitude à conduire des véhicules et à utiliser des machines
YAXWER n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
4.8 Effets indésirables
Résumé du profil de sécurité
Le profil général de sécurité de YAXWER est cohérent dans toutes les indications approuvées.
Une hypocalcémie a été très fréquemment rapportée suite à l’administration de denosumab, le plus souvent au cours des deux premières semaines suivant l’injection. L’hypocalcémie peut être sévère et symptomatique (voir rubrique 4.8 - Description d’effets indésirables sélectionnés). Les diminutions du calcium sérique étaient généralement gérées de manière appropriée par une supplémentation en calcium et en vitamine D. Les effets indésirables les plus fréquents associés au denosumab sont les douleurs musculo-squelettiques. Des cas d’ostéonécrose de la mâchoire (voir rubriques 4.4
et 4.8 - description d’effets indésirables sélectionnés) ont été fréquemment observés chez les patients traités par denosumab.
Tableau récapitulatif des effets indésirables
La convention suivante a été utilisée pour la classification des effets indésirables basée sur les taux d’incidence au cours de quatre études cliniques de phase III, de deux études de phase II et de l’expérience après commercialisation (voir Tableau 1) : très fréquent (≥ 1/10), fréquent (≥ 1/100,
< 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence et de classe de système d’organe, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1. Effets indésirables rapportés chez des patients présentant une affection maligne avancée avec atteinte osseuse, un myélome multiple ou une tumeur osseuse à cellules géantes
Classe MedDRA de système d’organe | Catégorie de fréquence | Effet indésirable |
Tumeurs bénignes, malignes et | Fréquent | Second cancer primitif1 |
Affections du système immunitaire | Rare | Hypersensibilité médicamenteuse1 |
Rare | Réaction anaphylactique1 | |
Troubles du métabolisme et de la nutrition | Très fréquent | Hypocalcémie1, 2 |
Fréquent | Hypophosphatémie | |
Peu fréquent | Hypercalcémie après l’arrêt du traitement chez les patients | |
Affections respiratoires, thoraciques et médiastinales | Très fréquent | Dyspnée |
Affections gastro-intestinales | Très fréquent | Diarrhée |
Fréquent | Extraction dentaire | |
Affections de la peau et du tissu sous-cutané | Fréquent | Hyperhidrose |
Peu fréquent | Éruptions lichénoïdes d’origine médicamenteuse1 | |
Affections musculo- squelettiques et du tissu conjonctif | Très fréquent | Douleur musculo-squelettique1 |
Fréquent | Ostéonécrose de la mâchoire1 | |
Peu fréquent | Fractures fémorales atypiques1 | |
Fréquence indéterminée | Ostéonécrose du conduit auditif externe3,4 |
1 Voir rubrique Description d’effets indésirables sélectionnés
2 Voir rubrique Autres populations particulières
3 Voir rubrique 4.4
4 Effet de classe
Description d’effets indésirables sélectionnés
Hypocalcémie
Une incidence plus élevée d’hypocalcémie a été observée chez les patients traités par le denosumab comparé à l’acide zolédronique dans les essais cliniques de prévention des complications osseuses (SRE : skeletal related events).
L’incidence la plus élevée d’hypocalcémie a été observée dans une étude de phase III chez des patients présentant un myélome multiple. Une hypocalcémie a été rapportée chez 16,9 % des patients traités par denosumab et chez 12,4 % des patients traités par l’acide zolédronique. Une diminution de la calcémie de grade 3 a été observée chez 1,4 % des patients traités par denosumab et chez 0,6 % des patients traités par l’acide zolédronique. Une diminution de la calcémie de grade 4 a été observée chez 0,4 % des patients traités par denosumab et chez 0,1 % des patients traités par l’acide zolédronique.
Lors de trois essais cliniques de phase III contrôlés contre comparateur actif menés chez des patients présentant une pathologie maligne avec atteinte osseuse, une hypocalcémie a été rapportée chez 9,6 % des patients traités par denosumab et chez 5,0 % des patients traités par l’acide zolédronique.
Une diminution de grade 3 de la calcémie a été observée chez 2,5 % des patients traités par denosumab et chez 1,2 % des patients traités par acide zolédronique. Une diminution de grade 4 de la calcémie a été
observée chez 0,6 % des patients traités par denosumab et chez 0,2 % des patients traités par acide zolédronique (voir rubrique 4.4).
Dans deux essais cliniques de phase II avec un bras unique menés chez des patients atteints de tumeurs osseuses à cellules géantes, une hypocalcémie a été rapportée chez 5,7 % des patients. Aucun des évènements indésirables n’a été considéré comme grave.
Des cas d’hypocalcémie symptomatique sévère (incluant des cas d’issue fatale) ont été rapportés après la commercialisation de YAXWER, la majorité des cas survenant durant les premières semaines d’initiation de la thérapie. Les exemples de manifestations cliniques d’hypocalcémie symptomatique sévère ont inclus allongement de l’intervalle du QT, tétanie, convulsions et altération de l’état mental (y compris coma) (voir rubrique 4.4). Les symptômes d’hypocalcémie survenus au cours des études cliniques incluaient des paresthésies ou raideurs musculaires, contractions, spasmes et crampes musculaires.
Ostéonécrose de la mâchoire (ONM)
Au cours des essais cliniques, l’incidence des ONM a été plus élevée lors des durées d’exposition plus longues ; des ONM ont également été diagnostiquées après l’arrêt du traitement par denosumab avec une majorité de cas se déclarant dans les 5 mois après la dernière dose. Les patients ayant des antécédents d’ONM ou d’ostéomyélite de la mâchoire, un problème dentaire ou à la mâchoire nécessitant une intervention chirurgicale, une chirurgie bucco-dentaire non cicatrisée ou toute intervention dentaire invasive planifiée ont été exclus des essais cliniques.
Lors des essais cliniques de prévention des SRE, une incidence plus élevée d’ONM a été observée chez les patients traités par le denosumab comparé à l’acide zolédronique. L’incidence la plus élevée d’ONM a été observée dans une étude de phase III chez des patients présentant un myélome multiple. Durant la phase de traitement en double aveugle de cette étude, les ONM ont été confirmées chez
5,9 % des patients traités par denosumab (exposition médiane de 19,4 mois ; extrêmes : 1 - 52 mois) et chez 3,2 % des patients traités par l’acide zolédronique. À la fin de la phase de traitement en double aveugle de cette étude, l’incidence ajustée exprimée en patient-année des ONM confirmées dans le groupe denosumab (exposition médiane de 19,4 mois ; extrêmes : 1 - 52 mois), était de 2,0 pour
100 patients-années pendant la première année de traitement, de 5,0 pendant la seconde année et de 4,5 par la suite. Le temps médian d’apparition d’ONM était de 18,7 mois (extrêmes : 1 - 44 mois).
Au cours des trois essais cliniques de phase III contrôlés contre comparateur actif menés chez des patients présentant une affection maligne avec atteinte osseuse, une ONM a été confirmée dans les premières phases de traitement chez 1,8 % des patients traités par denosumab (exposition médiane de 12,0 mois ; extrêmes : 0,1–40,5 mois) et chez 1,3 % des patients traités par acide zolédronique. Les caractéristiques cliniques de ces cas étaient comparables entre les groupes de traitement. La plupart des patients ayant présenté une ONM confirmée avaient des antécédents d’extraction dentaire, de mauvaise hygiène buccale et/ou d’utilisation d’un appareil dentaire (81 % dans les deux groupes de traitement). La plupart des patients recevaient ou avaient reçu une chimiothérapie.
Les essais chez les patients atteints d’un cancer du sein ou de la prostate comprenaient une phase d’extension de traitement par denosumab (exposition médiane globale de 14,9 mois ;
extrêmes : 0,1 - 67,2 mois). Une ONM a été confirmée chez 6,9 % des patients atteints d’un cancer du sein et d’un cancer de la prostate pendant la phase d’extension du traitement.
L’incidence globale ajustée exprimée en patient-année des ONM confirmées était de 1,1 pour
100 patients-années pendant la première année de traitement, de 3,7 pendant la seconde année et de 4,6 par la suite. Le temps médian d’apparition d’ONM était de 20,6 mois (extrêmes : 4 – 53 mois).
Une étude observationnelle, rétrospective, non randomisée menée chez 2 877 patients atteints d’un cancer traités par denosumab ou acide zolédronique en Suède, au Danemark et en Norvège a montré que le taux d'incidence à 5 ans des ONM confirmées sur le plan médical était de 5,7 % (IC 95 % : 4,4-7,3 ; durée médiane de suivi de 20 mois [extrêmes : 0,2 - 60]) dans une cohorte de patients recevant du denosumab et de 1,4 % (IC 95 % : 0,8-2,3 ; durée médiane de suivi de 13 mois [extrêmes : 0,1 - 60])
dans une cohorte de patients distincte recevant de l’acide zolédronique. Le taux d'incidence à cinq ans des ONM chez les patients passant de l’acide zolédronique au denosumab était de 6,6 % (IC 95 % :
4,2-10,0 ; durée médiane de suivi de 13 mois [extrêmes : 0,2 - 60]).
Dans une étude de phase III menée chez des patients atteints de cancer de la prostate non métastatique (population de patients dans laquelle le denosumab n’est pas indiqué), avec une durée d’exposition au traitement plus longue allant jusqu’à 7 ans, l’incidence ajustée exprimée en patient-année des ONM confirmées était de 1,1 pour 100 patients-années pendant la première année de traitement, de 3,0 pendant la seconde année et de 7,1 par la suite.
Dans une étude clinique de phase II en ouvert, à long terme, menée chez des patients atteints de tumeurs osseuses à cellules géantes (étude 6, voir rubrique 5.1), l’ONM était confirmée chez 6,8 % des patients, dont un adolescent (nombre médian de 34 doses ; extrêmes : 4 - 116). À la fin de l’étude, la durée médiane de l’étude incluant la phase de suivi de sécurité était de 60,9 mois
(extrêmes : 0 - 112,6 mois). L’incidence ajustée exprimée en patient-année des ONM confirmées était globalement de 1,5 pour 100 patients-années (de 0,2 pour 100 patients-années pendant la première année de traitement, de 1,5 pendant la seconde année, de 1,8 pendant la troisième année, de 2,1 pendant la quatrième année, de 1,4 pendant la cinquième année et de 2,2 par la suite). Le temps médian d’apparition de l'ONM était de 41 mois (extrêmes : 11 - 96 mois).
L’étude 7 a été menée pour continuer pendant 5 années supplémentaires ou plus le suivi des patients atteints de tumeurs osseuses à cellules géantes qui étaient traités dans l’étude 6. Une ONM a été signalée chez 6 patients (11,8 %) parmi les 51 patients exposés, avec un total médian de 42 doses de denosumab. Trois de ces cas d’ONM ont été confirmés sur le plan médical.
Réactions d’hypersensibilité liées au médicament
Après la mise sur le marché, des réactions d’hypersensibilité, incluant de rares cas de réactions anaphylactiques, ont été rapportées chez des patients recevant du denosumab.
Fractures atypiques du fémur
Globalement, dans le programme d’études cliniques, des fractures fémorales atypiques ont été rapportées peu fréquemment chez les patients traités par denosumab, et le risque augmentait avec la durée du traitement. Ces événements sont survenus en cours de traitement et jusqu’à 9 mois après l’arrêt du traitement (voir rubrique 4.4).
Dans le programme d’études cliniques sur les tumeurs osseuses à cellules géantes, des fractures fémorales atypiques ont été rapportées fréquemment chez les patients traités par denosumab. Dans l’étude 6, l’incidence des fractures fémorales atypiques confirmées a été de 0,95 % (5/526) chez les patients atteints de tumeurs osseuses à cellules géantes. Au cours de l’étude 7 de suivi, l’incidence des fractures fémorales atypiques confirmées a été de 3,9 % (2/51) chez les patients exposés au denosumab.
Douleur musculo-squelettique
Des douleurs musculo-squelettiques, y compris des cas sévères, ont été signalées chez des patients traités par du denosumab après la commercialisation. Dans les essais cliniques, les douleurs
musculo-squelettiques étaient très fréquentes dans le groupe denosumab et dans le groupe acide zolédronique. Les douleurs musculo-squelettiques ayant conduit à l’arrêt du traitement étaient peu fréquentes.
Second cancer primitif
Au cours des quatre essais cliniques de phase III contrôlés contre comparateur actif, et menés en double aveugle chez des patients présentant une affection maligne avancée avec atteinte osseuse, des cas de second cancer primitif ont été rapportés chez 1,5 % (54 sur 3 691) des patients traités par denosumab (exposition médiane de 13,8 mois ; extrêmes : 1,0 - 51,7) et 0,9 % (33 sur 3 688) des patients traités par acide zolédronique (exposition médiane de 12,9 mois ; extrêmes : 1,0 - 50,8).
L’incidence cumulée des cas de second cancer primitif à un an était de 1,1 % pour le denosumab et de 0,6 % pour l’acide zolédronique respectivement.
Aucun profil particulier de type de cancer ou de groupe de cancer relié au traitement n’a été mis en évidence.
Chez les patients atteints d’une tumeur osseuse à cellules géantes, l’incidence de second cancer, y compris de cancers des os ou autres cancers a été de 3,8 % (20/526) dans l’étude 6. Au cours de l’étude 7 de suivi, l’incidence a été de 11,8 % (6/51) chez les patients exposés au denosumab.
Éruptions lichénoïdes d’origine médicamenteuse
Des éruptions lichénoïdes d’origine médicamenteuse (par exemple des réactions de type lichen plan) ont été rapportées chez des patients après commercialisation.
Population pédiatrique
Le denosumab a été étudié au cours d’un essai clinique mené en ouvert incluant 28 adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes. Sur la base de ces données limitées, le profil des évènements indésirables semble comparable à celui observé chez les adultes.
Des hypercalcémies cliniquement significatives après l’arrêt du traitement ont été rapportées chez des enfants après commercialisation (voir rubrique 4.4).
Autres populations particulières
Insuffisance rénale
Dans une étude clinique menée chez des patients n’étant pas atteints de cancer avancé, insuffisants rénaux sévères (clairance de la créatinine < 30 mL/min) ou dialysés, le risque de développer une hypocalcémie était plus élevé en l’absence de supplémentation en calcium. Le risque de développer une hypocalcémie pendant le traitement par YAXWER augmente avec le degré d’insuffisance rénale. Au cours d’un essai clinique mené chez des patients ne présentant pas de cancer avancé, 19 % des patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 mL/min) et 63 % des patients dialysés ont développé une hypocalcémie malgré une supplémentation en calcium. L’incidence globale de l’hypocalcémie cliniquement significative était de 9 %.
Une augmentation de l’hormone parathyroïdienne a également été observée chez les patients insuffisants rénaux sévères ou dialysés, traités par denosumab. La surveillance de la calcémie et un apport adapté de calcium et de vitamine D sont particulièrement importants chez les patients insuffisants rénaux (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir Annexe V.
4.9 Surdosage
Aucun cas de surdosage n’a été rapporté au cours des essais cliniques. Le denosumab a été administré lors d’études cliniques à des doses allant jusqu’à 180 mg toutes les quatre semaines et 120 mg par semaine pendant trois semaines.
5. PROPRIÉTÉS PHARMACOLOGIQUES
5.1 Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Médicaments du traitement des maladies osseuses – Autres médicaments affectant la structure et la minéralisation de l’os, code ATC : M05BX04
YAXWER est un médicament biosimilaire. Des informations détaillées sont disponibles sur le site web de l'Agence européenne des médicaments http://www.ema.europa.eu.
Mécanisme d’action
RANKL est une protéine transmembranaire ou soluble essentielle pour la formation, la fonction et la survie des ostéoclastes, le seul type de cellules responsables de la résorption osseuse. Une augmentation de l’activité des ostéoclastes, stimulée par RANKL, est un médiateur majeur de la destruction osseuse dans les atteintes osseuses métastatiques et le myélome multiple. Le denosumab est un anticorps monoclonal humain (IgG2) qui cible RANKL et s’y lie avec une affinité et une spécificité élevées, s'opposant ainsi à l'interaction RANK/RANKL et réduisant le nombre et la fonction des ostéoclastes, diminuant de ce fait la résorption et la destruction osseuses induites par un cancer.
Les tumeurs osseuses à cellules géantes sont caractérisées par des cellules stromales néoplasiques exprimant RANK ligand et des cellules géantes de type ostéoclastique exprimant RANK. Chez les patients atteints de tumeurs osseuses à cellules géantes, le denosumab se lie au RANK ligand, et réduit ou élimine de façon significative les cellules géantes de type ostéoclastique. Par conséquent, l’ostéolyse diminue et le stroma de la tumeur proliférative est remplacé par de l’os non-prolifératif, différencié, à tissu dense.
Effets pharmacodynamiques
Dans les études cliniques de phase II menées chez des patients atteints de pathologie maligne avancée avec atteinte osseuse, l'administration sous-cutanée (SC) de denosumab toutes les 4 semaines (Q4W) ou toutes les 12 semaines a entraîné une réduction rapide des marqueurs de la résorption osseuse (uNTx/Cr, CTx sérique), avec des réductions médianes d'environ 80 % pour uNTx/Cr en l'espace d'une semaine, indépendamment d'un traitement antérieur par bisphosphonates ou du niveau initial d'uNTx/Cr. Dans les essais cliniques de phase III menés chez des patients atteints de tumeurs malignes avancées avec atteinte osseuse, des réductions médianes d'uNTx/Cr d'environ 80 % ont été maintenues pendant 49 semaines de traitement par le denosumab (120 mg toutes les 4 semaines).
Immunogénicité
Des anticorps anti-denosumab peuvent apparaître au cours du traitement par le denosumab. Aucune corrélation apparente entre l'apparition d'anticorps et la pharmacocinétique, la réponse clinique ou les effets indésirables n'a été observée.
Efficacité clinique et sécurité chez des patients présentant une tumeur solide avancée avec métastases osseuses
Trois études contrôlées, en double-aveugle et randomisées ont comparé l’efficacité et la sécurité de 120 mg de denosumab par voie sous-cutanée toutes les quatre semaines à celles de 4 mg d’acide zolédronique (dose adaptée en cas d’altération de la fonction rénale) par voie intraveineuse toutes les quatre semaines chez des patients présentant une pathologie maligne avancée avec atteinte osseuse et non encore traités par un bisphosphonate intraveineux : patients adultes atteints de cancer du sein (étude 1), d’autres tumeurs solides ou de myélome multiple (étude 2) et de cancer de la prostate résistant à la castration (étude 3). Parmi ces études contrôlées contre comparateur actif, la sécurité a été évaluée chez 5 931 patients. Les patients ayant des antécédents d’ONM ou d’ostéomyélite de la mâchoire, une affection dentaire ou de la mâchoire nécessitant une chirurgie buccale, une chirurgie bucco-dentaire non cicatrisée ou une intervention dentaire invasive prévue ont été exclus des essais cliniques. Les critères principaux et secondaires ont porté sur la survenue d’une ou plusieurs complications osseuses (SRE : skeletal related events). Dans les études démontrant la supériorité du denosumab sur l’acide zolédronique, une phase pré-spécifiée de 2 ans d’extension de traitement avec du denosumab en ouvert a été proposée aux patients. Un SRE est défini comme l’un des événements suivants : une fracture pathologique (vertébrale ou non vertébrale), une radiothérapie osseuse (incluant l’utilisation de radio-isotopes), une chirurgie osseuse ou une compression médullaire.
Le denosumab a réduit le risque de développer des complications osseuses uniques ou multiples (premières complications osseuses et suivantes) chez des patients atteints de tumeur solide avec atteinte osseuse (voir Tableau 2).
Tableau 2. Efficacité clinique chez des patients présentant une pathologie maligne avancée avec atteinte osseuse
| Étude 1 Cancer du sein | Étude 2 Autres tumeurs | Étude 3 Cancer de la prostate | Etude combinée Cancer avancé | ||||
| denosumab | Acide zolé- dronique | denosumab | Acide zolé- dronique | denosumab | Acide zolé- dronique | denosumab | Acide zolé- dronique |
N | 1 026 | 1 020 | 886 | 890 | 950 | 951 | 2 862 | 2 861 |
Première complication osseuse (SRE) | ||||||||
Délai médian (mois) | NA | 26,4 | 20,6 | 16,3 | 20,7 | 17,1 | 27,6 | 19,4 |
Différence des délais médians (mois) | ND | 4,2 | 3,5 | 8,2 | ||||
HR (IC95 %) | 0,82 (0,71 - 0,95)/18 | 0,84 | 0,82 | 0,83 | ||||
Valeur de p pour les | < 0,0001† / 0,0101† | 0,0007† / 0,0619† | 0,0002† / 0,0085† | < 0,0001 / < 0,0001 | ||||
% de patients | 30,7 | 36,5 | 31,4 | 36,3 | 35,9 | 40,6 | 32,6 | 37,8 |
Premier SRE et suivants* | ||||||||
Nombre moyen/ patient | 0,46 | 0,60 | 0,44 | 0,49 | 0,52 | 0,61 | 0,48 | 0,57 |
Rapport des taux (IC95 %) /RRR(%) | 0,77 (0,66 - 0,89) / | 0,90 (0,77 - 1,04) | 0,82 (0,71 - 0,94) / | 0,82 (0,75 - 0,89) / | ||||
Valeur de p pour le test de supériorité | 0,0012† | 0,1447† | 0,0085† | < 0,0001 | ||||
TME par an | 0,45 | 0,58 | 0,86 | 1,04 | 0,79 | 0,83 | 0,69 | 0,81 |
Premier SRE ou HCM | ||||||||
Délai médian (mois) | NA | 25,2 | 19,0 | 14,4 | 20,3 | 17,1 | 26,6 | 19,4 |
HR (IC95 %) / RRR(%) | 0,82 (0,70 - 0,95) / | 0,83 (0,71 - 0,97) / | 0,83 (0,72 - 0,96) / | 0,83 (0,76 - 0,90) / | ||||
Valeur de p pour le test de supériorité | 0,0074 | 0,0215 | 0,0134 | < 0,0001 | ||||
Première radiothérapie osseuse | ||||||||
Délai médian (mois) | NA | NA | NA | NA | NA | 28,6 | NA | 33,2 |
HR (IC95 %) / RRR(%) | 0,74 (0,59 - 0,94) / | 0,78 (0,63 - 0,97)/ | 0,78 (0,66 - 0,94)/ | 0,77 (0,69 - 0,87) / | ||||
Valeur de p pour le test de supériorité | 0,0121 | 0,0256 | 0,0071 | < 0,0001 | ||||
NA : non atteint ; ND : non disponible ; HCM : hypercalcémie maligne ; TME : Taux de Morbidité d’Evénement osseux ; HR : hazard ratio ; RRR : risque relatif réduit ; † Les valeurs de p corrigées sont présentées pour les études 1, 2 et 3 (critères premier SRE et premier SRE et suivants) ; *Tient compte de tous les évènements osseux au cours du temps ; seuls les événements survenus ≥ 21 jours après l’événement précédent sont pris en compte.
** inclus CPNPC, cancer du rein, cancer colorectal, cancer du poumon à petites cellules, cancer de la vessie, cancer de la tête et du cou, cancer GI/génito-urinaire et autres à l’exclusion des cancers du sein et de la prostate.
Figure 1. Courbe de Kaplan-Meier du délai de survenue du premier SRE au cours des études
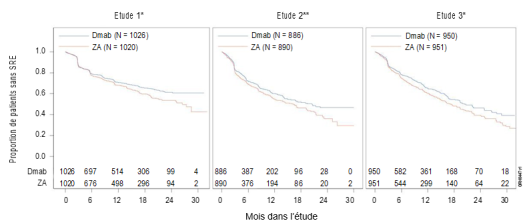
Dmab = Denosumab 120 mg Q4W ZA = Acide zolédronique 4 mg Q4W N = Nombre de patients randomisés
* = Supériorité statistiquement significative ; ** = Non-infériorité statistiquement significative
Progression de la maladie et survie globale chez les patients présentant une tumeur solide avec métastases osseuses
La progression de la maladie a été comparable entre les groupes denosumab et acide zolédronique au cours de chacune des trois études et dans l’analyse pré-spécifiée des trois études combinées.
Dans les études 1, 2 et 3, la survie globale a été comparable entre les patients présentant une pathologie maligne avec atteinte osseuse traités par denosumab ou par l’acide zolédronique : patientes atteintes de cancer du sein (hazard ratio et IC95 % : 0,95 [0,81 - 1,11]), patients atteints de cancer de la prostate (hazard ratio et IC95 % : 1,03 [0,91 - 1,17]) et patients atteints d’autres tumeurs solides ou de myélome multiple (hazard ratio et IC95 % : 0,95 [0,83 - 1,08]). Une analyse post hoc de l’étude 2 (patients atteints d’autres tumeurs solides ou de myélome multiple) a évalué la survie globale des patients présentant les trois types tumoraux utilisés pour la stratification (cancer du poumon non à petites cellules, myélome multiple, autre). La survie globale a été plus longue avec denosumab chez les patients atteints de cancer du poumon non à petites cellules (hazard ratio [IC95 %] : 0,79
[0,65 - 0,95] ; n = 702), plus longue avec l’acide zolédronique chez ceux atteints de myélome multiple (hazard ratio [IC95 %] : 2,26 [1,13 - 4,50] ; n = 180), et comparable avec denosumab et l’acide zolédronique chez les patients présentant un autre type tumoral (hazard ratio [IC95 %] : 1,08
[0,90 - 1,30] ; n = 894). Cette étude n’a pas pris en compte les facteurs pronostiques et les traitements anti-néoplasiques. Une analyse combinée pré spécifiée des études 1, 2 et 3 a indiqué que la survie globale avait été similaire entre les groupes denosumab et acide zolédronique (hazard ratio et IC95 % : 0,99 [0,91 - 1,07]).
Effet sur la douleur
Le délai d’amélioration de la douleur (soit une diminution ≥ 2 points du score BPI-SF de douleur augmentée par rapport à l’entrée dans l’étude) a été comparable entre les groupes denosumab et acide zolédronique de chaque étude et dans l’analyse intégrée. Dans une analyse post hoc des données combinées, le délai médian d’aggravation de la douleur (score de douleur augmentée > 4 points) chez
les patients ayant une douleur légère ou aucune douleur à l'entrée dans l’étude, a été plus long avec YAXWER qu’avec l’acide zolédronique (198 jours contre 143) (p = 0,0002).
Efficacité clinique chez les patients présentant un myélome multiple
Le denosumab a été évalué dans une étude internationale, randomisée (1:1), en double-aveugle, contrôlée comparant le denosumab à l’acide zolédronique chez des patients atteints d’un myélome multiple nouvellement diagnostiqué, étude 4.
Dans cette étude, 1 718 patients présentant un myélome multiple avec au moins une lésion osseuse ont été randomisés pour recevoir une administration sous-cutanée de 120 mg de denosumab toutes les
4 semaines (Q4W) ou une administration intraveineuse (IV) de 4 mg d’acide zolédronique toutes les 4 semaines (dose ajustée à la fonction rénale). Le critère d’évaluation principal était le délai de survenue du premier SRE en analyse de non-infériorité comparé à l’acide zolédronique. Les critères d’évaluation secondaires incluaient le délai de survenue du premier SRE en analyse de supériorité, le délai de survenue du premier SRE et des suivants en analyse de supériorité, et la survie globale. Un SRE est défini comme l’un des événements suivants : une fracture pathologique (vertébrale ou non vertébrale), une radiothérapie osseuse (incluant l’utilisation de radio-isotopes), une chirurgie osseuse ou une compression médullaire.
Parmi les deux bras de l’étude, 54,5 % des patients devaient recevoir une transplantation autologue de cellules souches périphériques, 95,8 % des patients ont reçu / devaient recevoir un nouvel agent du traitement du myélome (les thérapies nouvelles incluaient le bortézomib, le lénalidomide ou le thalidomide) en traitement de première ligne, et 60,7 % des patients avaient un antécédent de SRE. Le nombre de patients parmi les deux bras de l’étude avec un ISS (système international de stadification) de stade I, stade II et stade III au diagnostic était de 32,4 %, 38,2 % et 29,3 % respectivement.
Le nombre médian de doses administrées était de 16 pour le denosumab et de 15 pour l’acide zolédronique. Les résultats d’efficacité de l’étude 4 sont présentés en figure 2 et tableau 3.
Figure 2. Courbe de Kaplan-Meier du délai de survenue du premier SRE chez les patients atteints d’un myélome multiple nouvellement diagnostiqué
1,0 |
|
|
|
|
|
![]()
![]()
![]()
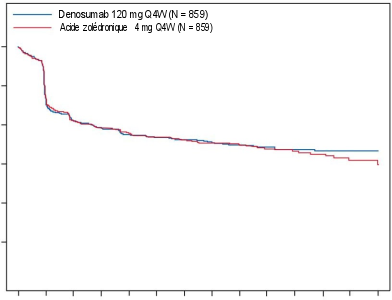 Denosumab 120 mg Q4W Acide zolédronique 4 mg Q4W
Denosumab 120 mg Q4W Acide zolédronique 4 mg Q4W
Mois dans l’étude
N = nombre de patients randomisés
Tableau 3. Résultats d’efficacité de YAXWER comparé à l’acide zolédronique chez les patients atteints d’un myélome multiple nouvellement diagnostiqué
| Denosumab (N = 859) | Acide zolédronique (N = 859) |
Premier SRE | ||
Nombre de patients qui avaient des SRE(%) | 376 (43,8) | 383 (44,6) |
Délai médian de survenue du SRE (mois) | 22,8 (14,7 ; NE) | 23,98 (16,56 ; |
Hazard ratio (IC 95 %) | 0,98 (0,85 ; 1,14) | |
| ||
Premier SRE et suivants | ||
Nombre moyen d’évènements/patient | 0,66 | 0,66 |
Rapport des taux (IC 95 %) | 1,01 (0,89 ; 1,15) | |
Taux de morbidité osseuse annuelle | 0,61 | 0,62 |
| ||
Premier SRE ou HCM | ||
Délai médian (mois) | 22,14 (14,26 ; NE) | 21,32 (13,86 ; 29,7) |
Hazard ratio (IC 95 %) | 0,98 (0,85 ; 1,12) | |
| ||
Première radiothérapie osseuse | ||
Hazard ratio (IC 95 %) | 0,78 (0,53 ; 1,14) | |
| ||
Survie globale | ||
Hazard ratio (IC 95 %) | 0,90 (0,70 ; 1,16) | |
NE : non estimable
HCM : hypercalcémie maligne
Efficacité clinique et sécurité chez les adultes et les adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes
La sécurité et l’efficacité du denosumab ont été étudiées lors de deux études en ouvert de phase II, avec un bras unique (études 5 et 6), menées chez 554 patients atteints de tumeurs osseuses à cellules géantes non résécables ou pour lesquelles la résection chirurgicale pouvait être associée à une morbidité sévère et une étude prospective de phase IV, multicentrique, menée en ouvert (étude 7) qui a fourni un suivi à long terme de la sécurité chez les patients ayant terminé l’étude 6. Les patients ont reçu une injection sous-cutanée de 120 mg de denosumab toutes les 4 semaines avec une dose supplémentaire de 120 mg les jours 8 et 15. Les patients ayant arrêté le denosumab sont ensuite entrés dans la phase de suivi de sécurité pour une durée minimale de 60 mois.
Pendant qu’ils étaient dans la phase de suivi de sécurité, un retraitement par denosumab était autorisé chez les patients qui avaient initialement démontré une réponse au traitement par denosumab (par exemple, en cas de récidive de la maladie).
Dans l’étude 5, 37 patients adultes atteints d’une tumeur osseuse à cellules géantes non résécable ou récurrente, confirmée par histologie, ont été inclus. Le critère d’évaluation principal pour cette étude était le taux de réponse, défini soit comme une élimination d’au moins 90 % des cellules géantes par rapport à l’inclusion (ou l’élimination totale des cellules géantes dans les cas où ces dernières représentaient < 5 % des cellules tumorales), soit comme l'absence de progression des lésions cibles d’après les mesures radiographiques dans les cas où l’histopathologie n’était pas disponible. Parmi les 35 patients inclus dans les analyses d’efficacité, 85,7 % (IC95 % : 69,7 ; 95,2) ont présenté une réponse au traitement par denosumab. Les 20 patients (100 %) avec évaluation histologique répondaient aux critères de réponse. Sur les 15 patients restants, 10 (67 %) d’entre eux évalués radiographiquement n’ont montré aucune progression des lésions cibles.
Dans l’étude 6, 535 adultes ou adolescents à maturité osseuse atteints de tumeurs osseuses à cellules géantes ont été inclus. Parmi ces patients, 28 étaient âgés de 12 à 17 ans. Les patients ont été répartis dans une des trois cohortes : la cohorte 1 comprenait les patients atteints de maladie chirurgicalement incurable (par exemple, des lésions sacrées, médullaires ou multiples, dont des métastases pulmonaires) ; la cohorte 2 comprenait les patients atteints de maladie chirurgicalement curable dont la chirurgie planifiée était associée à une morbidité sévère (par exemple, résection articulaire, amputation d’un membre ou hémipelvectomie) ; la cohorte 3 comprenait les patients ayant participé auparavant à l’étude 5 et ayant poursuivi dans cette étude. L’objectif principal était d’évaluer le profil de sécurité du denosumab chez les patients atteints de tumeurs osseuses à cellules géantes. Les critères d’évaluation secondaires pour cette étude comprenaient le temps de progression de la maladie (d’après l’évaluation de l’investigateur) pour la cohorte 1 et la proportion de patients n’ayant pas été opérés à 6 mois pour la cohorte 2.
Dans la cohorte 1, lors de l’analyse finale, 28 des 260 patients traités (10,8 %) ont présenté une progression de la maladie. Dans la cohorte 2, 219 des 238 patients évaluables (92,0 % ; IC 95 % : 87,8 %, 95,1 %) traités par denosumab n’avaient pas subi d’intervention chirurgicale à 6 mois. Sur les 239 patients de la cohorte 2 présentant une lésion cible à l’inclusion ou lors de l’étude qui n’était pas située dans les poumons ou les tissus mous, 82 patients au total (34,3 %) ont pu éviter une intervention chirurgicale pendant l’étude. Dans l’ensemble, les résultats concernant l’efficacité chez les adolescents à maturité squelettique étaient similaires à ceux observés chez les adultes.
L’étude 7 comptait 85 patients adultes qui avaient participé à l’étude 6 et qui l’avaient terminée. Les patients étaient autorisés à recevoir un traitement par denosumab pour leurs tumeurs osseuses à cellules géantes et tous les patients ont bénéficié d’un suivi sur 5 ans. L’objectif principal était d’évaluer le profil de sécurité à long terme du denosumab chez les patients atteints de tumeurs osseuses à cellules géantes.
Effet sur la douleur
Dans les cohortes 1 et 2 combinées de l’analyse finale, une réduction cliniquement significative de la douleur la plus sévère (c’est-à-dire une diminution de > 2 points par rapport à l'inclusion) a été rapportée chez 30,8 % des patients à risque (c’est-à-dire ceux qui avaient un score de douleur à l'inclusion le plus sévère ≥ 2 points) après une semaine de traitement, et ≥ 50 % à la semaine 5. Ces améliorations de la douleur ont été maintenues lors de toutes les évaluations suivantes.
Population pédiatrique
L'Agence européenne du médicament a dispensé de l'obligation de soumettre les résultats des études menées avec le médicament de référence contenant du denosumab dans toutes les sous-populations pédiatriques dans la prévention des complications osseuses chez les patients atteints de métastases osseuses et des sous-populations pédiatriques âgées de moins de 12 ans dans le traitement des tumeurs osseuses à cellules géantes (voir la rubrique 4.2 pour des informations sur l'utilisation pédiatrique).
Lors de l’étude 6, le denosumab a été évalué dans une sous-population de 28 patients adolescents (âgés de 13 à 17 ans) atteints de tumeurs osseuses à cellules géantes qui avaient atteint leur maturité squelettique, définie par au moins un os long mature (par exemple, la plaque de croissance apophysaire de l’humérus fermée) et un poids corporel ≥ 45 kg. Un patient adolescent atteint de maladie chirurgicalement incurable (N = 14) a présenté une récidive de la maladie lors du traitement initial. Treize des 14 patients atteints de maladie chirurgicalement curable dont la chirurgie planifiée était associée à une morbidité sévère n’avaient pas subi d’intervention chirurgicale à 6 mois.
5.2 Propriétés pharmacocinétiques
Absorption
Après l’administration sous-cutanée, la biodisponibilité a été de 62 %.
Biotransformation
Le denosumab est composé uniquement d'acides aminés et de carbohydrates, comme l'immunoglobuline native, et ne devrait pas être éliminé par métabolisme hépatique. Son métabolisme et son élimination devraient suivre les voies de clairance de l'immunoglobuline pour aboutir à une dégradation en petits peptides et en acides aminés individuels.
Elimination
Chez les patients atteints d’un cancer avancé, ayant reçu des doses multiples de 120 mg toutes les quatre semaines, une accumulation multipliant les concentrations sériques du denosumab par un facteur de 2 environ a été observée et l’état d’équilibre a été obtenu à six mois, traduisant une pharmacocinétique indépendante du temps. Chez les patients atteints de myélome multiple ayant reçu 120 mg toutes les 4 semaines, les concentrations résiduelles médianes variaient de moins de 8 % entre le 6ème et le 12ème mois. Chez les patients atteints de tumeurs osseuses à cellules géantes ayant reçu 120 mg toutes les 4 semaines avec une dose supplémentaire aux jours 8 et 15, l’état d’équilibre a été atteint au cours du premier mois de traitement. Entre la semaine 9 et la semaine 49, les concentrations résiduelles médianes variaient de moins de 9 %. La demi-vie moyenne a été de 28 jours (extrêmes : 14 - 55 jours) chez les patients ayant arrêté le traitement par 120 mg de denosumab toutes les quatre semaines.
Une analyse de la pharmacocinétique n’a révélé aucune modification cliniquement significative de l’exposition systémique au denosumab à l’état d’équilibre en fonction de l’âge (18 à 87 ans), de l’origine ethnique (africaine, hispanique, asiatique, et caucasienne), du sexe ou du type de tumeur solide ou de patients atteints de myélome multiple. Un poids corporel plus élevé a été associé à une exposition systémique plus faible et inversement. Cette tendance n'est toutefois pas considérée comme cliniquement pertinente puisque les effets pharmacodynamiques, basés sur les marqueurs du remodelage osseux ont été cohérents sur un large éventail de poids corporel.
Linéarité/non-linéarité
La pharmacocinétique du denosumab a été non linéaire par rapport à la dose sur une large gamme de doses, mais l’exposition a augmenté de façon quasi dose-dépendante à partir de 60 mg (ou 1 mg/kg). La non-linéarité est probablement due à la saturation de la voie d’élimination à faibles concentrations.
Insuffisance rénale
Lors d’études avec le denosumab (60 mg, n = 55 et 120 mg, n = 32) chez des patients ne présentant pas d’affection maligne avancée mais divers degrés d’altération de la fonction rénale, dont des patients sous dialyse, le degré d’insuffisance rénale n’avait aucun effet sur les paramètres pharmacocinétiques du denosumab ; l’ajustement posologique chez les patients insuffisants rénaux n’est pas requis. Une surveillance de la fonction rénale n’est pas nécessaire au cours du traitement par denosumab.
Insuffisance hépatique
Aucune étude spécifique n’a été menée chez des patients insuffisants hépatiques. En règle générale, les anticorps monoclonaux ne sont pas éliminés par des mécanismes de métabolisme hépatique. La présence d’une insuffisance hépatique ne devrait pas affecter la pharmacocinétique du denosumab.
Sujet âgé
Aucune différence globale portant sur la sécurité ou l’efficacité n’a été observée entre des patients âgés et plus jeunes. Des études cliniques contrôlées menées avec du denosumab chez des patients âgés de plus de 65 ans présentant une pathologie maligne avancée avec atteinte osseuse ont indiqué une efficacité et une sécurité comparables à celles observées chez des patients plus jeunes. Aucune adaptation posologique n’est nécessaire chez les patients âgés.
Population pédiatrique
Chez les adolescents à maturité squelettique (âgés de 12 à 17 ans) atteints de tumeurs osseuses à cellules géantes ayant reçu 120 mg toutes les 4 semaines avec une dose supplémentaire les jours 8 et 15, les paramètres pharmacocinétiques du denosumab étaient similaires à ceux observés chez les patients adultes atteints de tumeurs osseuses à cellules géantes.
5.3 Données de sécurité préclinique
L’activité biologique du denosumab chez l’animal étant spécifique aux primates non humains, l’évaluation de souris knockout obtenues par génie génétique ou l'administration d’autres inhibiteurs biologiques de la voie RANK/RANKL, par exemple OPG-Fc et RANK-Fc, ont été utilisés afin d’évaluer les propriétés pharmacodynamiques du denosumab dans des modèles de rongeurs.
L’OPG-Fc a réduit des lésions ostéolytiques, ostéoblastiques et ostéolytiques/ostéoblastiques, a retardé la formation de métastases osseuses de novo et a réduit la croissance de tumeurs osseuses dans des modèles murins de métastases osseuses de cancers du sein humains positifs ou négatifs pour les récepteurs aux estrogènes, de cancer de la prostate ou de cancer du poumon non à petites cellules.
L’association de l’OPG-Fc à un traitement hormonal (tamoxifène) ou à une chimiothérapie (docétaxel) dans ces modèles, a inhibé de façon additive la croissance de tumeurs osseuses secondaires à un cancer du sein, et de la prostate ou du poumon respectivement. Le RANK-Fc a réduit la prolifération hormono-induite dans l’épithélium mammaire et retardé la formation de tumeurs dans un modèle d’induction de tumeurs mammaires chez la souris.
Aucun test standard du potentiel génotoxique du denosumab n’a été réalisé, ces tests n’étant pas pertinents pour cette molécule. Toutefois, compte-tenu de sa nature, il est peu probable que le denosumab présente un quelconque potentiel génotoxique.
Le potentiel carcinogène du denosumab n'a pas été évalué dans le cadre d'études à long terme chez l'animal.
Dans des études de toxicité à doses unique ou répétées conduites chez des singes cynomolgus, des doses de denosumab entraînant une exposition systémique de 2,7 à 15 fois plus élevée que celle induite par la dose humaine recommandée n'ont eu aucun impact sur la physiologie cardiovasculaire, la fertilité des mâles ou des femelles et n'ont entraîné aucune toxicité spécifique sur les organes cibles.
Dans une étude menée chez le singe cynomolgus exposé au denosumab pendant une période correspondant au premier trimestre de la grossesse, des expositions systémiques au denosumab jusqu'à 9 fois plus élevées que l'exposition à la dose humaine recommandée, n'ont pas entraîné de toxicité maternelle, ni d'effet délétère pour le fœtus pendant une période correspondante au premier trimestre bien que les ganglions lymphatiques des fœtus n’aient pas été examinés.
Dans une autre étude menée chez le singe cynomolgus exposé pendant la gestation au denosumab, à des expositions systémiques 12 fois supérieures à celle de la dose humaine, ont été observés : une augmentation de mort-nés et de la mortalité postnatale, une croissance osseuse anormale aboutissant à une diminution de la résistance osseuse, une diminution de l’hématopoïèse, un alignement anormal des dents, une absence de ganglions lymphatiques périphériques et une diminution de la croissance néonatale. La dose sans effet indésirable sur la reproduction n’a pu être établie. Six mois après la naissance, les modifications osseuses étaient réversibles et aucun effet sur la poussée dentaire n’a été observé. Toutefois, les effets sur les ganglions lymphatiques et l’alignement anormal des dents ont persisté, et chez un animal, une minéralisation faible à modérée a été observée dans de nombreux tissus (la relation avec le traitement est incertaine). Aucune souffrance maternelle avant le travail n’a été observée et les effets indésirables maternels pendant le travail ont été peu fréquents. Le développement des glandes mammaires était normal.
Dans les études précliniques évaluant la qualité de l'os, conduites chez des singes traités au long cours par le denosumab, la diminution du remodelage osseux a été associée à une amélioration de la résistance osseuse et à une histologie osseuse normale.
Chez des souris mâles génétiquement modifiées pour exprimer hu-RANKL (souris knock-in) et sujettes à une fracture transcorticale, le denosumab a retardé la résorption du cartilage et le remodelage du cal fracturaire en comparaison au contrôle, mais la résistance biomécanique n'a pas été altérée.
Dans des études précliniques, une absence de lactation due à l'inhibition de la maturation de la glande mammaire (développement lobulo-alvéolaire au cours de la gestation) et l’altération de la formation des ganglions lymphatiques ont été observées chez des souris knockout n’exprimant pas RANK ou RANKL. Des souris knockout de RANK/RANKL en période néonatale ont présenté une diminution du poids corporel, une réduction de la croissance osseuse, une altération des cartilages de croissance et une absence de poussée dentaire. Une réduction de la croissance osseuse ainsi qu'une altération des cartilages de croissance et de la poussée dentaire ont été également observées lors d'études menées chez des rats ayant reçu un inhibiteur de RANKL en période néonatale ; ces modifications ont été partiellement réversibles à l’arrêt de ce traitement. De jeunes primates traités par des doses de denosumab de 2,7 et 15 fois (10 et 50 mg/kg) celles correspondant à l’exposition clinique ont présenté une anomalie des cartilages de croissance. Le traitement par le denosumab peut donc altérer la croissance osseuse chez les enfants avec des cartilages de croissance non soudés et peut inhiber la poussée dentaire.
6. DONNÉES PHARMACEUTIQUES
6.1 Liste des excipients
Acide acétique glacial*
Hydroxyde de sodium (pour ajustement du pH)* Sorbitol (E420)
Polysorbate 20 (E432)
Eau pour préparations injectables
* Le tampon acétate est formé par mélange d’acide acétique et d’hydroxyde de sodium.
6.2 Incompatibilités
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
6.3 Durée de conservation
3 ans.
Une fois sorti du réfrigérateur, YAXWER peut être conservé à température ambiante (jusqu’à 25 °C) pendant 30 jours maximum dans son emballage d'origine et son emballage extérieur afin de le protéger de la lumière. Il doit être utilisé dans la limite de cette période de 30 jours.
6.4 Précautions particulières de conservation
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
6.5 Nature et contenu de l’emballage extérieur
1,7 mL de solution en flacon en verre transparent (verre de type I selon la Ph. Eur.) scellé par un bouchon en caoutchouc bromobutyle recouvert d’une couche fluorée et d’un bouchon plastique à opercule avec joint en aluminium.
Boîtes de un, trois ou quatre flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6 Précautions particulières d’élimination et manipulation
- Avant administration, la solution YAXWER doit être inspectée visuellement. Ne pas injecter la solution si elle contient des particules ou si elle est trouble, ou présente un changement de coloration.
- Ne pas agiter.
- Afin d'éviter une gêne au point d'injection, laisser le flacon atteindre la température ambiante (jusqu’à 25 °C) avant l’injection et injecter lentement.
- Tout le contenu du flacon doit être injecté.
- Une aiguille de 27 G est recommandée pour l'administration de denosumab.
- Le flacon ne doit pas être ponctionné une seconde fois.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/25/1934/001
EU/1/25/1934/002
EU/1/25/1934/003
9. DATE DE PREMIÈRE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
Date de première autorisation :
10. DATE DE MISE A JOUR DU TEXTE
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l'Agence européenne des médicaments http://www.ema.europa.eu/.
ANNEXE II
A. FABRICANTS DE LA SUBSTANCE ACTIVE D’ORIGINE BIOLOGIQUE ET FABRICANTS RESPONSABLES DE LA LIBÉRATION DES LOTS
B. CONDITIONS OU RESTRICTIONS DE DÉLIVRANCE ET D’UTILISATION
C. AUTRES CONDITIONS ET OBLIGATIONS DE L’AUTORISATION DE MISE SUR LE MARCHÉ
D. CONDITIONS OU RESTRICTIONS EN VUE D’UNE UTILISATION SÛRE ET EFFICACE DU MÉDICAMENT
A. FABRICANT DE LA SUBSTANCE ACTIVE D'ORIGINE BIOLOGIQUE ET FABRICANTS RESPONSABLES DE LA LIBÉRATION DES LOTS
Nom et adresse du fabricant de la substance active d’origine biologique
Chemical Works of Gedeon Richter Plc.
(Gedeon Richter Plc.)
Richter Gedeon utca 20.
4031 Debrecen
Hongrie
Nom et adresse des fabricants responsables de la libération des lots
Chemical Works of Gedeon Richter Plc.
(Gedeon Richter Plc.)
Richter Gedeon utca 20.
4031 Debrecen
Hongrie
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
Le nom et l’adresse du fabricant responsable de la libération du lot concerné doivent figurer sur la notice du médicament.
B. CONDITIONS OU RESTRICTIONS DE DÉLIVRANCE ET D’UTILISATION
Médicament soumis à prescription médicale restreinte (voir Annexe I : Résumé des Caractéristiques du Produit, rubrique 4.2).
C. AUTRES CONDITIONS ET OBLIGATIONS DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Rapports périodiques actualisés de sécurité (PSURs)
Les exigences relatives à la soumission des PSURs pour ce médicament sont définies dans la liste des dates de référence pour l’Union (liste EURD) prévue à l’article 107 quater, paragraphe 7, de la directive 2001/83/CE et ses actualisations publiées sur le portail web européen des médicaments.
D. CONDITIONS OU RESTRICTIONS EN VUE D’UNE UTILISATION SÛRE ET EFFICACE DU MÉDICAMENT
E.
Plan de gestion des risques (PGR)
Le titulaire de l’autorisation de mise sur le marché réalise les activités de pharmacovigilance et interventions requises décrites dans le PGR adopté et présenté dans le Module 1.8.2 de l’autorisation de mise sur le marché, ainsi que toutes actualisations ultérieures adoptées du PGR.
De plus, un PGR actualisé doit être soumis :
- à la demande de l’Agence européenne des médicaments ;
- dès lors que le système de gestion des risques est modifié, notamment en cas de réception de nouvelles informations pouvant entraîner un changement significatif du profil bénéfice/risque, ou lorsqu’une étape importante (pharmacovigilance ou minimisation du risque) est franchie.
Mesures additionnelles de minimisation du risque
Le titulaire de l’autorisation de mise sur le marché doit s’assurer de la mise en place d’une carte d’information au patient concernant les ostéonécroses de la mâchoire.
ANNEXE III ÉTIQUETAGE ET NOTICE
A. ÉTIQUETAGE
MENTIONS DEVANT FIGURER SUR L’EMBALLAGE EXTÉRIEUR
CARTON
Yaxwer 120 mg solution injectable
denosumab
Chaque flacon contient 120 mg de denosumab dans 1,7 mL de solution (70 mg/ml).
Acide acétique glacial, hydroxyde de sodium (pour ajustement du pH), sorbitol (E420), polysorbate 20, eau pour préparations injectables.
Solution injectable
1 flacon à usage unique
3 flacons à usage unique
4 flacons à usage unique
Lire la notice avant utilisation.
Voie sous-cutanée.
Ne pas agiter.
QR code à inclure
www.yaxwerinfo.com
Tenir hors de la vue et de la portée des enfants.
A usage unique.
EXP
A conserver au réfrigérateur.
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l’abri de la lumière.
10. PRÉCAUTIONS PARTICULIÈRES D’ÉLIMINATION DES MÉDICAMENTS NON UTILISÉS OU DES DÉCHETS PROVENANT DE CES MÉDICAMENTS S’IL Y A LIEU
11. NOM ET ADRESSE DU TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
12. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/25/1934/001 1 flacon à usage unique
EU/1/25/1934/002 3 flacon à usage unique
EU/1/25/1934/003 4 flacon à usage unique
13. NUMÉRO DU LOT
Lot
14. CONDITIONS DE PRESCRIPTION ET DE DÉLIVRANCE
15. INDICATIONS D’UTILISATION
16. INFORMATIONS EN BRAILLE
Yaxwer
17. IDENTIFIANT UNIQUE - CODE-BARRES 2D
code-barres 2D portant l’identifiant unique inclus.
18. IDENTIFIANT UNIQUE - DONNÉES LISIBLES PAR LES HUMAINS
PC
SN
NN
MENTIONS MINIMALES DEVANT FIGURER SUR LES PETITS CONDITIONNEMENTS PRIMAIRES
ÉTIQUETAGE DU FLACON
Yaxwer 120 mg solution injectable
denosumab
SC
EXP
Lot
1,7 mL
B. NOTICE
Notice : information du patient
YAXWER 120 mg solution injectable
denosumab
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Vous pouvez y contribuer en signalant tout effet indésirable que vous observez. Voir en fin de rubrique 4 comment déclarer les effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Vous pouvez y contribuer en signalant tout effet indésirable que vous observez. Voir en fin de rubrique 4 comment déclarer les effets indésirables.
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
- Gardez cette notice. Vous pourriez avoir besoin de la relire.
- Si vous avez d’autres questions, interrogez votre médecin, ou pharmacien.
- Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
- Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
- Votre médecin vous donnera une carte d’information au patient, qui contient des informations de sécurité importantes dont vous devrez avoir connaissance avant et pendant votre traitement par YAXWER.
YAXWER contient du denosumab, une protéine (anticorps monoclonal) qui ralentit la destruction osseuse due à un cancer se disséminant dans les os (métastases osseuses) ou d’une tumeur osseuse à cellules géantes.
YAXWER est utilisé afin de prévenir des complications osseuses graves dues à des métastases osseuses (ex. : fracture, compression de la moelle épinière ou nécessité de radiothérapie ou chirurgie) chez l’adulte atteint de cancer avancé.
YAXWER est également utilisé dans le traitement des tumeurs osseuses à cellules géantes qui ne peuvent pas être traitées par chirurgie ou lorsque la chirurgie n’est pas la meilleure option, chez les adultes et les adolescents ayant terminé leur croissance.
- Si vous êtes allergique au denosumab ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Votre professionnel de santé ne devra pas vous traiter par YAXWER si votre taux de calcium dans le sang est très bas et n’a pas été traité.
Votre professionnel de santé ne devra pas vous traiter par YAXWER si vous présentez des plaies non cicatrisées résultant d’une chirurgie bucco-dentaire.
Avertissements et précautions
Adressez-vous à votre médecin avant de recevoir YAXWER.
Supplémentation en calcium et vitamine D
Vous devez prendre un supplément de calcium et de vitamine D au cours de votre traitement par YAXWER, sauf si le taux de calcium dans votre sang est élevé. Votre médecin s’en entretiendra avec vous. Si le taux de calcium dans votre sang est faible, votre médecin peut décider de vous prescrire un supplément de calcium avant que vous ne commenciez votre traitement par YAXWER.
Faible taux de calcium dans le sang
Prévenez immédiatement votre médecin si vous ressentez au cours de votre traitement par YAXWER des spasmes, des contractions ou des crampes dans vos muscles et/ou un engourdissement ou des picotements dans vos doigts, vos orteils ou autour de votre bouche et/ou si vous présentez des convulsions, une confusion ou une perte de conscience. Le taux de calcium dans votre sang peut être bas.
Insuffisance rénale
Prévenez votre médecin si vous avez ou avez eu des problèmes rénaux sévères, si vous êtes ou avez été atteint d’une insuffisance rénale ou si vous êtes sous dialyse, ce qui pourrait augmenter le risque d’abaissement du taux de calcium dans votre sang, particulièrement si vous ne prenez pas un supplément de calcium.
Complications au niveau de la bouche, des dents ou des mâchoires
Un effet indésirable appelé ostéonécrose de la mâchoire (altération des os de la mâchoire) a fréquemment été rapporté (pouvant affecter jusqu’à 1 personne sur 10) chez les patients recevant des injections de denosumab pour des affections liées au cancer. L’ostéonécrose de la mâchoire peut aussi apparaître après l’arrêt du traitement.
Il est important d’essayer de prévenir l’apparition de l’ostéonécrose de la mâchoire car c’est une affection qui peut être douloureuse et difficile à traiter. Afin de réduire le risque de développer une ostéonécrose de la mâchoire, voici quelques précautions à prendre :
- Avant de recevoir le traitement, prévenez votre médecin/infirmier (professionnel de santé) si vous avez des affections au niveau de la bouche ou des dents. Votre médecin devra retarder l’initiation de votre traitement si vous présentez des plaies non cicatrisées dans la bouche, résultant de traitements dentaires ou d’une chirurgie bucco-dentaire. Votre médecin pourra alors vous recommander un examen dentaire avant de commencer le traitement par YAXWER.
- Pendant le traitement, vous devrez maintenir une bonne hygiène bucco-dentaire et faire des bilans dentaires réguliers. Si vous portez des prothèses dentaires, vous devrez vous assurer qu’elles sont bien ajustées.
- Si vous êtes en cours de traitement dentaire ou si vous avez prévu une intervention de chirurgie dentaire (par exemple extraction dentaire), informez votre médecin de votre traitement dentaire et votre dentiste de votre traitement par YAXWER.
- Prévenez immédiatement votre médecin ou votre dentiste si vous ressentez des problèmes au niveau de la bouche ou des dents tels que le déchaussement d’une dent, une douleur ou un gonflement, un ulcère non cicatrisé ou un écoulement, car cela pourrait être le signe d’une ostéonécrose de la mâchoire.
Le risque de développer une ostéonécrose de la mâchoire peut être plus élevé chez les patients traités par chimiothérapie et/ou radiothérapie, par corticoïdes ou par un inhibiteur de l’angiogenèse (utilisé dans le traitement du cancer), ayant subi une chirurgie dentaire, ne recevant pas de soins dentaires réguliers, ayant une affection des gencives et chez les fumeurs.
Fractures inhabituelles de l’os de la cuisse
Certaines personnes ont développé des fractures inhabituelles de l’os de la cuisse lorsqu’elles étaient traitées par denosumab. Contactez votre médecin si vous ressentez une douleur nouvelle ou inhabituelle au niveau de la hanche, de l’aine ou de la cuisse.
Taux élevés de calcium dans le sang après l’arrêt du traitement par YAXWER
Certains patients atteints de tumeurs osseuses à cellules géantes ont développé des taux élevés de calcium dans le sang quelques semaines à quelques mois après l’arrêt du traitement. Votre médecin vous examinera pour détecter les signes et symptômes de taux élevés de calcium après l’arrêt de votre traitement par YAXWER.
Enfants et adolescents
YAXWER ne doit pas être utilisé chez les enfants et les adolescents de moins de 18 ans, excepté chez les adolescents atteints de tumeurs osseuses à cellules géantes qui ont terminé leur croissance. YAXWER n’a pas été étudié chez l’enfant et l’adolescent atteints d’autres cancers s’étant propagés dans l’os.
Autres médicaments et YAXWER
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris un médicament obtenu sans ordonnance. Il est particulièrement important d’informer votre médecin si vous êtes traité(e) par
- un autre médicament contenant du denosumab
- un bisphosphonate.
Vous ne devez pas prendre YAXWER avec un autre médicament contenant du denosumab ou des bisphosphonates.
Grossesse et allaitement
Si vous êtes enceinte ou si vous allaitez, si vous pensez être enceinte ou si vous envisagez d'avoir un enfant, demandez conseil à votre médecin avant de prendre ce médicament.
YAXWER n’a pas été étudié chez la femme enceinte. Il est important de prévenir votre médecin si vous êtes enceinte ou pensez l’être ou si vous prévoyez de débuter une grossesse. YAXWER ne doit pas être utilisé chez la femme enceinte. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement par YAXWER et pendant au moins 5 mois après l’arrêt du traitement par YAXWER.
Si vous débutez une grossesse pendant le traitement par YAXWER ou moins de 5 mois après l’arrêt de votre traitement par YAXWER, vous devez en informer votre médecin.
On ne sait pas si YAXWER est excrété dans le lait maternel. Si vous allaitez ou envisagez d’allaiter, il est important de le signaler à votre médecin. Votre médecin vous aidera ainsi à décider si vous devez cesser d’allaiter ou interrompre le traitement par YAXWER, en tenant compte du bénéfice de l’allaitement pour le nourrisson et celui du traitement par YAXWER pour la mère.
Si vous allaitez pendant le traitement par YAXWER, vous devez en informer votre médecin. Conduite de véhicules et utilisation de machines
YAXWER n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
YAXWER contient du sorbitol, du polysorbate 20 et du sodium
Ce médicament contient 78 mg de sorbitol par dose (1,7 mL).
Ce médicament contient 0,17 mg de polysorbate 20 par dose (1,7 mL). Les polysorbates peuvent provoquer des réactions allergiques. Informez votre médecin si vous avez ou si votre enfant a des allergies connues.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose (1,7 mL), c.-à-d. qu’il est essentiellement « sans sodium ».
YAXWER doit être administré sous la responsabilité d’un professionnel de santé.
La dose recommandée de YAXWER est de 120 mg administrés une fois toutes les quatre semaines au moyen d’une seule injection sous la peau (sous-cutanée). YAXWER sera injecté dans votre cuisse, votre abdomen ou le haut de votre bras. Si vous êtes traité(e) pour une tumeur osseuse à cellules géantes, vous recevrez une dose supplémentaire 1 semaine et 2 semaines après la dose initiale.
Ne pas agiter.
Vous devez également prendre un supplément de calcium et de vitamine D pendant votre traitement par YAXWER, sauf si vous avez un excès de calcium dans le sang. Votre médecin en parlera avec vous.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Prévenez immédiatement votre médecin si vous ressentez l’un des symptômes suivants pendant votre traitement par YAXWER (peut affecter plus d’une personne sur 10) :
- Spasmes, contractions, crampes dans vos muscles, engourdissement ou picotements dans vos doigts, vos orteils ou autour de votre bouche et/ou convulsions, confusion ou perte de conscience. Ces symptômes peuvent être dus à un faible taux de calcium dans le sang. Un taux de calcium sanguin bas peut être à l’origine d’une modification du rythme cardiaque appelée allongement du QT, qui peut être confirmé par électrocardiogramme (ECG).
Prévenez immédiatement votre médecin ou votre dentiste si vous ressentez l’un des symptômes suivants pendant votre traitement par YAXWER ou après l’arrêt du traitement (peut affecter jusqu’à 1 personne sur 10) :
- Douleurs persistantes et/ou des gonflements ou des ulcères non cicatrisants dans la bouche et/ou les mâchoires, écoulement, engourdissement ou sensation de lourdeur des mâchoires, déchaussement d’une dent pouvant être dus à une altération des os de la mâchoire (ostéonécrose).
Effets indésirables très fréquents (pouvant affecter plus d’une personne sur 10) :
- Douleur osseuse, articulaire et/ou musculaire, qui peut être parfois sévère,
- Essoufflement,
- Diarrhée.
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
- Faible taux de phosphates dans le sang (hypophosphatémie),
- Extraction d’une dent,
- Sueurs excessives,
- Chez les patients atteints d’un cancer avancé : développement d’une autre forme de cancer.
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
- Taux élevés de calcium dans le sang (hypercalcémie) après l’arrêt du traitement chez les patients atteints de tumeurs osseuses à cellules géantes,
- Douleur nouvelle ou inhabituelle au niveau de la hanche, de l’aine ou de la cuisse (ceci peut être un signe précoce d’une possible fracture de l’os de la cuisse),
- Éruptions pouvant survenir sur la peau ou ulcères dans la bouche (éruptions lichénoïdes d’origine médicamenteuse).
Effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000) :
- Réactions allergiques (ex. respiration sifflante ou difficultés à respirer ; gonflement du visage, des lèvres, de la langue, de la gorge et d’autres parties du corps ; éruptions, démangeaisons ou urticaire de la peau). Dans de rares cas les réactions allergiques peuvent être sévères.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
- Consultez votre médecin si vous présentez des douleurs de l’oreille, des écoulements de l’oreille et/ou une infection de l’oreille. Il pourrait s’agir de signes de lésions osseuses de l’oreille.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien, ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration décrit en Annexe V. En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois
A conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
Le flacon peut être laissé hors du réfrigérateur pour atteindre la température ambiante (jusqu’à 25 °C) avant l’injection. Cela rendra l’injection plus confortable.
Une fois sorti du réfrigérateur, YAXWER peut être conservé à température ambiante (jusqu’à 25 °C) pendant 30 jours maximum dans son emballage d'origine et son emballage extérieur afin de le protéger de la lumière. Il doit être utilisé au cours de cette période de 30 jours.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
- La substance active est le denosumab. Chaque flacon contient 120 mg de denosumab dans 1,7 mL de solution (correspondant à 70 mg/mL).
- Les autres composants sont : acide acétique glacial, hydroxyde de sodium, sorbitol (E420), polysorbate 20 (E432) et eau pour préparations injectables.
Comment se présente YAXWER et contenu de l’emballage extérieur
YAXWER est une solution injectable.
YAXWER est une solution injectable limpide et incolore à légèrement jaune, sans particules visibles.
1,7 mL de solution en flacon en verre transparent (verre de type I selon la Ph. Eur.) scellé par un bouchon en caoutchouc bromobutyle recouvert d’une couche fluorée et d’un bouchon plastique à opercule avec joint en aluminium.
Chaque boîte contient un, trois ou quatre flacons à usage unique. Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’Autorisation de mise sur le marché
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
Fabricants
Chemical Works of Gedeon Richter Plc.
(Gedeon Richter Plc.)
Richter Gedeon utca 20.
4031 Debrecen
Hongrie
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
Cette notice a été révisée la dernière fois le .
Autres sources d'information
Des informations détaillées sur ce produit sont également disponibles en scannant le QR code inclus ci-dessous ou sur le conditionnement extérieur à l'aide d'un smartphone. Les mêmes informations sont également disponibles sur l'URL suivante : www.yaxwerinfo.com
QR Code à inclure
Des informations détaillées sur ce médicament sont disponibles sur le site web de l'Agence européenne des médicaments : http://www.ema.europa.eu, et sur le site web de {nom de l'agence de l'État membre (lien)}.
L'information suivante est destinée aux professionnels de santé :
- Avant l'administration, la solution YAXWER doit être inspectée visuellement. Ne pas injecter la solution si elle contient des particules visibles, si elle est trouble, ou si elle présente un changement de coloration.
- Ne pas agiter.
- Afin de limiter la douleur au point d'injection, laisser le flacon atteindre la température ambiante (jusqu'à 25 °C) avant d'injecter et injecter lentement.
- Tout le contenu du flacon doit être injecté.
- Une aiguille de 27 G est recommandée pour l'administration du denosumab.
- Le flacon ne doit pas être ponctionné une seconde fois.
Tout produit inutilisé ou déchets doivent être éliminés conformément à la règlementation en vigueur.
PRIX
| Code CNK | Emballage | Prix | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|
| 4947180 | YAXWER 70MG/ML SOL INJ FL 1,7ML | € 161,13 | Oui | € 12,8 | € 8,5 |
| 4947206 | YAXWER 70MG/ML SOL INJ FL 4X1,7ML | € 611,62 | Oui | € 12,8 | € 8,5 |