SAMENVATTING VAN DE PRODUCTKENMERKEN
1. NAAM VAN HET GENEESMIDDEL
ALPROLIX 250 I.E. poeder en oplosmiddel voor oplossing voor injectie
ALPROLIX 500 I.E. poeder en oplosmiddel voor oplossing voor injectie
ALPROLIX 1000 I.E. poeder en oplosmiddel voor oplossing voor injectie
ALPROLIX 2000 I.E. poeder en oplosmiddel voor oplossing voor injectie
ALPROLIX 3000 I.E. poeder en oplosmiddel voor oplossing voor injectie
2. KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
ALPROLIX 250 I.E. poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 250 I.E. humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX bevat na reconstitutie ongeveer 250 I.E. (50 I.E./ml) humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX 500 I.E. poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 500 I.E. humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX bevat na reconstitutie ongeveer 500 I.E. (100 I.E./ml) humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX 1000 I.E. poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 1000 I.E. humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX bevat na reconstitutie ongeveer 1000 I.E. (200 I.E./ml) humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX 2000 I.E. poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 2000 I.E. humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX bevat na reconstitutie ongeveer 2000 I.E. (400 I.E./ml) humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX 3000 I.E. poeder en oplosmiddel voor oplossing voor injectie
Elke injectieflacon bevat nominaal 3000 I.E. humane stollingsfactor IX (rDNA), eftrenonacog alfa.
ALPROLIX bevat na reconstitutie ongeveer 3000 I.E. (600 I.E./ml) humane stollingsfactor IX (rDNA), eftrenonacog alfa.
De sterkte (I.E.) wordt bepaald aan de hand van de one‑stage clotting‑test (eentrapsstollingstest) van de Europese Farmacopee. De specifieke activiteit van ALPROLIX is 55‑84 I.E./mg eiwit.
Eftrenonacog alfa (recombinant‑humane‑stollingsfactor IX, Fc‑fusie‑eiwit (rFIXFc)) heeft 867 aminozuren. Het is een factorproduct met hoge zuiverheid dat wordt geproduceerd met recombinant‑DNA‑technologie in een cellijn van humane embryonale niercellen (HEK) zonder toevoeging van exogene eiwitten die van mensen of dieren afkomstig zijn, in de celkweek, de zuivering of de eindformulering.
Hulpstof met bekend effect
0,3 mmol (6,4 mg) natrium per injectieflacon.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
3. FARMACEUTISCHE VORM
Poeder en oplosmiddel voor oplossing voor injectie.
Poeder: gelyofiliseerd, wit tot gebroken wit poeder of cake.
Oplosmiddel: heldere tot kleurloze oplossing.
pH: 6,5 tot 7,5
Osmolaliteit: 255 tot 345 mOsm/kg
4. KLINISCHE GEGEVENS
4.1 Therapeutische indicaties
Behandeling en profylaxe van bloedingen bij patiënten met hemofilie B (aangeboren factor IX‑deficiëntie).
ALPROLIX kan worden gebruikt voor alle leeftijdsgroepen.
4.2 Dosering en wijze van toediening
Behandeling moet plaatsvinden onder het toezicht van een arts met ervaring in de behandeling van hemofilie.
Monitoring van de behandeling
Tijdens behandeling wordt een toepasselijke bepaling van de factor IX‑waarde aanbevolen als leidraad voor de toe te dienen dosis en de frequentie van herhaalde injecties. De respons op factor IX kan van patiënt tot patiënt verschillen, waardoor verschillende halfwaardetijden en verschillende recoverywaarden worden aangetoond. Een dosis die wordt gebaseerd op het lichaamsgewicht moet mogelijk worden aangepast bij patiënten met onder‑ of overgewicht. Met name in het geval van grote chirurgische ingrepen is een precieze monitoring van de substitutietherapie aan de hand van stollingsanalyse (factor IX‑activiteit in plasma) onontbeerlijk.
Wanneer een one‑stage clotting assay in vitro op basis van de geactiveerde partiële tromboplastinetijd (aPTT) wordt gebruikt voor bepaling van factor IX‑activiteit in bloedmonsters van patiënten, kunnen resultaten van factor IX‑activiteit in plasma aanzienlijk beïnvloed worden door zowel het type aPTT‑reagens als de referentiestandaard die in de test worden gebruikt. Dit is vooral van belang wanneer het laboratorium en/of het in de test gebruikte reagens worden gewijzigd.
Metingen met een one‑stage clotting assay met gebruikmaking van een aPTT‑reagens op basis van kaoline zullen waarschijnlijk resulteren in een onderschatting van activiteitniveau.
Dosering
Dosering en duur van de substitutietherapie zijn afhankelijk van de ernst van de factor IX‑deficiëntie, van de plaats en ernst van de bloeding en van de klinische toestand van de patiënt.
Het aantal eenheden van factor IX dat toegediend wordt, wordt volgens de huidige WHO‑standaard voor factor IX‑producten uitgedrukt in Internationale Eenheden (I.E.). De activiteit van factor IX in plasma wordt uitgedrukt als een percentage (t.o.v. normaal humaan plasma) of in Internationale Eenheden (t.o.v. een internationale norm voor factor IX in plasma).
Eén Internationale Eenheid (I.E.) van recombinant‑factor IX Fc‑activiteit is gelijk aan de hoeveelheid factor IX in één ml normaal humaan plasma.
Behandeling naar behoefte (on demand)
De berekening van de vereiste dosis van recombinant‑factor IX Fc is gebaseerd op de empirische vaststelling dat 1 Internationale Eenheid (I.E.) factor IX per kg lichaamsgewicht de factor IX‑activiteit in plasma met 1% van de normale activiteit (I.E./dl) verhoogt. De vereiste dosis wordt bepaald met behulp van de volgende formule:
Benodigd aantal eenheden = lichaamsgewicht (kg) × gewenste factor IX‑toename (%) (I.E./dl) × {reciprook van waargenomen herstel (I.E./kg per I.E./dl)}
De toe te dienen hoeveelheid en de toedieningsfrequentie moeten altijd afgestemd worden op de klinische werkzaamheid voor die specifieke persoon. Indien een dosis moet worden herhaald om de bloeding onder controle te brengen, moet rekening worden gehouden met de langere halfwaardetijd van ALPROLIX (zie rubriek 5.2). De verwachting is dat de tijd tot piekactiviteit niet vertraagd zal zijn.
In het geval van de volgende bloedingsepisodes mag de factor IX‑activiteit niet lager zijn dan de vermelde plasma‑activiteit (in % van de normale waarde of I.E./dl) in de betreffende periode. Tabel 1 kan gebruikt worden als leidraad bij de dosering tijdens bloedingsepisodes en chirurgische ingrepen:
Tabel 1: Leidraad voor dosering van ALPROLIX bij behandeling van bloedingsepisodes en chirurgische ingrepen
Mate van hemorragie / type chirurgische ingreep | Vereiste factor IX-waarde (%) (I.E./dl) | Doseringsfrequentie (uren) / duur van behandeling (dagen) |
Hemorragie |
|
|
Hemartrose, spierbloeding of orale bloeding in een vroeg stadium | 20‑40 | Injectie elke 48 uur herhalen, tot de bloedingsepisode zoals aangegeven door pijn is verdwenen of genezing is bereikt. |
Grotere hemartrose, spierbloeding of hematoom | 30‑60 | Injectie elke 24 tot 48 uur herhalen tot pijn en acute invaliditeit zijn verdwenen. |
Levensbedreigende bloedingen | 60‑100 | Injectie elke 8 tot 24 uur herhalen tot de toestand niet meer levensbedreigend is. |
Chirurgische ingrepen |
|
|
Kleine operatie, waaronder tandextractie | 30‑60 | Injectie na 24 uur herhalen, naar behoefte tot genezing is bereikt1. |
Grote chirurgische ingrepen | 80‑100 | Injectie elke 8 tot 24 uur als nodig herhalen tot adequate wondgenezing, vervolgens behandeling gedurende ten minste nog eens 7 dagen voortzetten om een factor IX‑activiteit van 30% tot 60% (I.E./dl) te handhaven. |
1 Bij sommige patiënten en onder sommige omstandigheden kan het doseringsinterval worden verlengd tot 48 uur (zie rubriek 5.2 voor farmacokinetische gegevens).
Profylaxe
Voor een langdurige profylaxe tegen bloedingen, zijn de aanbevolen startschema’s ofwel:
- 50 I.E./kg eenmaal per week, dosis aanpassen op basis van de individuele respons of
- 100 I.E./kg eenmaal elke 10 dagen, interval aanpassen op basis van de individuele respons. Sommige patiënten bij wie de bloedingen goed onder controle zijn op een schema van eenmaal elke 10 dagen, kunnen worden behandeld met een interval van 14 dagen of langer.
De hoogste aanbevolen dosis voor profylaxe is 100 I.E./kg.
Ouderen
Er is beperkte ervaring bij patiënten ≥ 65 jaar.
Pediatrische patiënten
Voor kinderen jonger dan 12 jaar zijn mogelijk hogere of frequentere doses vereist en de aanbevolen startdosis is 50‑60 I.E./kg elke 7 dagen. Voor adolescenten van 12 jaar en ouder zijn de dosisaanbevelingen dezelfde als voor volwassenen. Zie rubriek 5.1 en 5.2.
De hoogste aanbevolen dosis voor profylaxe is 100 I.E./kg.
Wijze van toediening
Intraveneus gebruik.
In geval van zelftoediening of toediening door een zorgverlener is de juiste training noodzakelijk.
ALPROLIX moet over een periode van enkele minuten intraveneus worden geïnjecteerd. De toedieningssnelheid moet worden afgestemd op wat de patiënt als prettig ervaart en mag niet hoger zijn dan 10 ml/min.
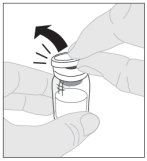
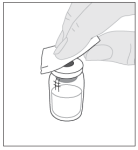
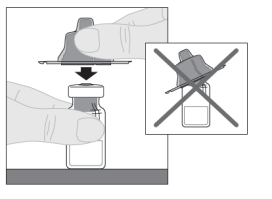
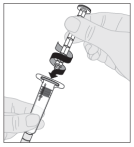
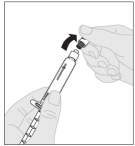
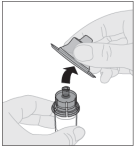
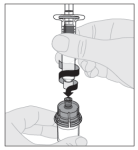
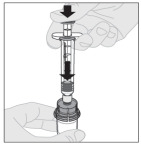
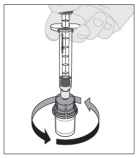
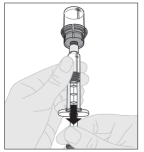
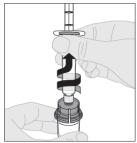
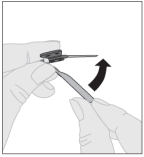
Voor instructies over reconstitutie van het geneesmiddel voorafgaand aan toediening, zie rubriek 6.6.
4.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor een van de in rubriek 6.1 vermelde hulpstoffen.
4.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Overgevoeligheids‑ of allergische reacties (die kunnen bestaan uit angio‑oedeem, brandend en stekend gevoel op de infuusplaats, koude rillingen, overmatig blozen, gegeneraliseerde urticaria, hoofdpijn, netelroos, hypotensie, lethargie, nausea, rusteloosheid, tachycardie, beklemd gevoel in de borst, tintelingen, braken, piepende ademhaling) zijn zelden waargenomen en kunnen in sommige gevallen verergeren tot ernstige anafylaxie (waaronder shock). In enkele gevallen ontwikkelden deze reacties zich tot ernstige anafylaxie en traden deze reacties op terwijl er zich vrijwel tegelijkertijd remmers tegen factor IX ontwikkelden (zie ook rubriek 4.4). Nefrotisch syndroom is gemeld na een poging tot inductie van immuuntolerantie bij hemofilie B‑patiënten met factor IX‑remmers en een voorgeschiedenis van allergische reactie.
Patiënten met hemofilie B kunnen neutraliserende antilichamen (remmers) tegen factor IX ontwikkelen. Indien dergelijke remmers voorkomen, zal de toestand zich manifesteren als een onvoldoende klinische respons. In dergelijke gevallen wordt aangeraden contact op te nemen met een gespecialiseerd hemofiliecentrum.
Er bestaat een potentieel risico op trombo‑embolische episodes na de toediening van factor IX‑producten, waarbij het risico groter is voor preparaten met geringe zuiverheid. Het gebruik van factor IX‑producten met geringe zuiverheid is in verband gebracht met gevallen van myocardinfarct, gedissemineerde intravasale stolling, veneuze trombose en longembolie. Het gebruik van factor IX met hoge zuiverheid is zelden in verband gebracht met trombo‑embolische complicaties.
Lijst van bijwerkingen in tabelvorm
Eerder behandelde patiënten: in totaal werden 153 patiënten met ernstige hemofilie B geobserveerd in klinische fase III‑onderzoeken en een verlengingsonderzoek. Ongewenste voorvallen zijn waargenomen over in totaal 561 persoonjaren. Het totale aantal blootstellingsdagen was 26.106 met een mediaan van 165 (bereik 1 tot 528) blootstellingsdagen per persoon.
Niet eerder behandelde patiënten: in totaal werden 33 patiënten met ernstige hemofilie B geobserveerd in één klinisch onderzoek. Ongewenste voorvallen zijn waargenomen over in totaal 57,51 persoonjaren. Het totale aantal blootstellingsdagen was 2.233 met een mediaan van 76 (bereik 1 tot 137) blootstellingsdagen per persoon.
Tabel 2 hieronder is in overeenstemming met de classificatie van de systeem/orgaanklasse volgens MedDRA (niveau van SOC en voorkeursterm).
Frequenties zijn geëvalueerd in overeenstemming met de volgende afspraak: zeer vaak (≥ 1/10); vaak (≥ 1/100, < 1/10); soms (≥ 1/1.000, < 1/100); zelden (≥ 1/10.000, < 1/1.000); zeer zelden (< 1/10.000); niet bekend (kan met de beschikbare gegevens niet worden bepaald). In de tabel worden de bijwerkingen vermeld die zijn gemeld in de klinische onderzoeken en die zijn vastgesteld bij postmarketinggebruik.
Tabel 2: Bijwerkingen gemeld voor ALPROLIX
Systeem/orgaanklasse volgens MedDRA | Bijwerkingen | Frequentiecategorie |
Bloed‑ en lymfestelselaandoeningen | Factor IX-remming | Vaak1 |
Immuunsysteemaandoeningen | Overgevoeligheid | Vaak1 |
Voedings‑ en stofwisselingsstoornissen | Verminderde eetlust | Soms |
Zenuwstelselaandoeningen | Hoofdpijn | Vaak |
Hartaandoeningen | Hartkloppingen | Soms |
Bloedvataandoeningen | Hypotensie | Soms |
Maagdarmstelselaandoeningen | Paresthesie oraal | Vaak |
Nier‑ en urinewegaandoeningen | Obstructieve uropathie | Vaak |
Algemene aandoeningen en toedieningsplaatsstoornissen | Erytheem op injectieplaats | Vaak |
1 De frequentie is gebaseerd op optreden in het onderzoek bij niet eerder behandelde patiënten. Beide voorvallen van factor IX-remming en overgevoeligheid traden op bij één niet eerder behandelde patiënt in onderzoek IV. Zie Beschrijving van geselecteerde bijwerkingen.
Beschrijving van geselecteerde bijwerkingen
Gedurende het gehele klinische onderzoeksprogramma was er één (niet eerder behandelde) patiënt in onderzoek IV die een lage-titerfactor IX-remmer ontwikkelde in combinatie met overgevoeligheid (zie rubriek 5.1). Na het in de handel brengen zijn ontwikkeling van en overgevoeligheid (waaronder anafylaxie) voor factor IX‑remmers waargenomen.
Pediatrische patiënten
Frequentie, type en ernst van bijwerkingen bij kinderen zijn naar verwachting dezelfde als bij volwassenen. Zie rubriek 5.1 voor omvang en leeftijdstypering van de veiligheidsdatabase bij kinderen.
Melding van vermoedelijke bijwerkingen
Het is belangrijk om na toelating van het geneesmiddel vermoedelijke bijwerkingen te melden. Op deze wijze kan de verhouding tussen voordelen en risico’s van het geneesmiddel voortdurend worden gevolgd. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via:
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten
Afdeling Vigilantie
Galileelaan 5/03 | Postbus 97 |
Website: www.eenbijwerkingmelden.be
e-mail: adr@fagg.be

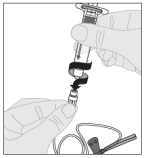
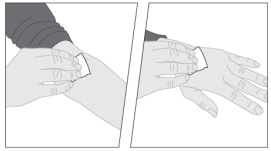 2.
2. 
7. HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Swedish Orphan Biovitrum AB (publ)
SE‑112 76 Stockholm
Zweden
8. NUMMER(S) VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU/1/16/1098/001
EU/1/16/1098/002
EU/1/16/1098/003
EU/1/16/1098/004
EU/1/16/1098/005
10. DATUM VAN HERZIENING VAN DE TEKST
05/01/2024
Gedetailleerde informatie over dit geneesmiddel is beschikbaar op de website van het Europees Geneesmiddelenbureau http://www.ema.europa.eu.
PRIJZEN
| CNK code | Verpakking | ATC5 code | Prijs | Af-fabriek prijs | Voorschriftplichtig |
|---|---|---|---|---|---|
| 3456597 | ALPROLIX 250IE PDR+SOLV OPL INJ 1 FL + SER PR.5ML | B02BD04 | € 311,83 | - | Ja |
| 3456605 | ALPROLIX 500IE PDR+SOLV OPL INJ 1 FL + SER PR.5ML | B02BD04 | € 613,1 | - | Ja |
| 3456613 | ALPROLIX 1000IE PDR+SOLV OPL INJ 1 FL + SER PR.5ML | B02BD04 | € 1215,64 | - | Ja |
| 3456621 | ALPROLIX 2000IE PDR+SOLV OPL INJ 1 FL + SER PR.5ML | B02BD04 | € 2420,7 | - | Ja |
| 3456639 | ALPROLIX 3000IE PDR+SOLV OPL INJ 1 FL + SER PR.5ML | B02BD04 | € 3625,76 | - | Ja |