RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
JEMPERLI 500 mg solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un flacon de 10 ml de solution à diluer pour perfusion contient 500 mg de dostarlimab.
Chaque ml de solution à diluer pour perfusion contient 50 mg de dostarlimab.
Le dostarlimab est un anticorps monoclonal (AcM) humanisé (immunoglobuline G4 [IgG4]) anti-PD-1 (programmed cell death protein-1), produit par la technique de l’ADN recombinant dans des cellules de mammifères, d’ovaire de hamster chinois (CHO).
Excipients à effet notoire
2mg de polysorbate 80 par unité de dose
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution à diluer pour perfusion (stérile).
Solution transparente à légèrement opalescente, incolore à jaune, essentiellement sans particules visibles.
La solution à diluer pour perfusion a un pH d’environ 6,0 et une osmolalité d’environ 300 mOsm/kg.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
JEMPERLI est indiqué en association avec le carboplatine et le paclitaxel pour le traitement de première ligne des patientes adultes atteintes d’un cancer de l’endomètre (CE) avancé nouvellement diagnostiqué ou récidivant et candidates à un traitement systémique.
JEMPERLI est indiqué en monothérapie pour le traitement des patientes adultes atteintes d’un cancer de l’endomètre, récidivant ou avancé, qui présente une déficience du système de réparation des mésappariements des bases (dMMR)/ une instabilité microsatellitaire élevée (MSI-H) en progression après ou pendant une chimiothérapie à base de platine.
4.2 Posologie et mode d’administration
Le traitement doit être initié et supervisé par des médecins spécialistes qualifiés et expérimentés dans le traitement du cancer.
La détermination du statut dMMR/MSI-H de la tumeur doit être faite en utilisant une méthode validée telles que IHC, PCR ou NGS * (voir rubrique 5.1 pour les informations sur les tests utilisés dans les essais cliniques).
*IHC=immunohistochimie ; PCR= réaction en chaine par polymérase ; NGS= séquençage de nouvelle génération
Posologie
JEMPERLI en association avec le carboplatine et le paclitaxel
Lorsque JEMPERLI est administré en association avec le carboplatine et le paclitaxel, se référer aux informations de prescription complètes des produits individuels (voir aussi la rubrique 5.1).
La dose recommandée est de 500 mg de dostarlimab toutes les 3 semaines en association avec le carboplatine et le paclitaxel toutes les 3 semaines pendant 6 cycles suivi de 1000 mg de dostarlimab en monothérapie toutes les 6 semaines pour tous les cycles suivants.
Le schéma posologique en association avec le carboplatine et le paclitaxel est présenté dans le Tableau 1.
![]()
Tableau 1. Schéma posologique pour JEMPERLI en association avec le carboplatine et le paclitaxel
| 500 mg une fois toutes les 3 semaines |
| 1000 mg une fois toutes les 6 semaines en monothérapie jusqu’à progression de la maladie ou toxicité inacceptable , ou pour une durée maximale allant jusqu’à 3 ans (1 Cycle = 6 semaines) | ||||||||
Cycle | Cycle 1 | Cycle 2 | Cycle 3 | Cycle 4 | Cycle 5 | Cycle 6 | Cycle 7 | Cycle 8 | Cycle 9 |
| |
Semaine | 1 | 4 | 7 | 10 | 13 | 16 | 19 | 25 | 31 | ||
3 semaines entre le Cycle 6 et le Cycle 7
a Administrer le dostarlimab avant le carboplatine et le paclitaxel le même jour.
L’administration de dostarlimab doit se poursuivre selon le calendrier recommandé jusqu’à progression de la maladie ou toxicité inacceptable, ou pour une durée allant jusqu’à 3 ans (voir rubrique 5.1).
JEMPERLI en monothérapie
La dose recommandée en monothérapie est de 500 mg de dostarlimab toutes les 3 semaines pour les 4 premiers cycles, puis de 1 000 mg toutes les 6 semaines pour tous les cycles suivants.
Le schéma posologique en monothérapie est présenté dans le Tableau 2.
Tableau 2. Schéma posologique pour JEMPERLI en monothérapie![]()
![]()
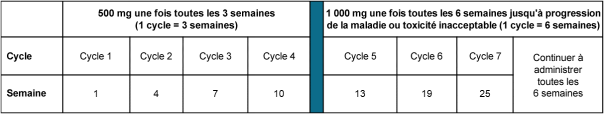
3 semaines entre le cycle 4 et le cycle 5
L’administration de dostarlimab doit se poursuivre selon le calendrier recommandé jusqu’à progression de la maladie ou toxicité inacceptable (voir rubrique 5.1).
Modifications de la dose
Réduire la dose n’est pas recommandé. Il peut être nécessaire de retarder l’administration ou d’arrêter le traitement si le profil de sécurité et de tolérance observé individuellement le requiert. Les modifications recommandées pour la gestion des effets indésirables sont précisées dans le tableau 3.
Les recommandations détaillées pour la prise en charge des effets indésirables d’origine immunologique et des réactions liées à la perfusion sont décrites à la rubrique 4.4.
Tableau 3. Recommandations pour la suspension ou l’arrêt de JEMPERLI | ||
Effets indésirables d’origine immunologique | Grade de sévéritéa | Modification de traitement |
Colite | 2 ou 3 | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes de toxicité sont revenus aux grades 0 ou 1. |
4 | Arrêter définitivement le traitement. | |
Hépatite | Grade 2 avec ASATb ou ALATc > 3 et jusqu’à 5 x LSNd | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes de toxicité sont revenus aux grades 0 ou 1. |
Grade ≥ 3 avec ASAT ou ALAT> 5x LSN ou bilirubine totale > 3 x LSN | Arrêter définitivement le traitement (voir exception ci-dessous) e | |
Diabète sucré de type 1 (DT1) | 3 ou 4 (hyperglycémie) |
|
Hypophysite ou insuffisance surrénalienne | 2, 3 ou 4 | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes sont revenus au grade 0 ou 1. Arrêter définitivement le traitement en cas de récidive ou d’aggravation malgré une hormonothérapie adéquate. |
Hypothyroïdie ou hyperthyroïdie | | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes sont revenus au grade 0 ou 1. |
Pneumopathie inflammatoire | 2 | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes sont revenus aux grades 0 ou 1. Arrêter définitivement le traitement si la patiente revient à une sévérité de grade 2. |
3 ou 4 | Arrêter définitivement le traitement. | |
Néphrite | 2 | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes sont revenus au grade 0 ou 1. |
3 ou 4 | Arrêter définitivement le traitement. | |
Réactions dermatologiques exfoliatives (par exemple SJSf, NETg, DRESSh) | Suspecté | Suspendre le traitement pour tout grade. Reprendre l’administration du traitement si réaction non confirmée et lorsque les symptômes sont revenus au grade 0 ou 1. |
Confirmé | Arrêter définitivement le traitement. | |
Myocardite | 2,3 ou 4 | Arrêter définitivement le traitement. |
Toxicités neurologiques sévères (syndrome myasthénique/myasthénie grave, syndrome de Guillain-Barré, encéphalite, myélite transverse) | 2,3 ou 4 | Arrêter définitivement le traitement. |
Autres effets indésirables d’origine immunologique (y compris mais sans s’y limiter : myosites, sarcoïdoses, anémies hémolytiques auto-immunes, pancréatites, iridocyclites, uvéites, cétoacidoses diabétiques, arthralgie, rejets de greffes d’organes solides, réaction du greffon contre l’hôte | 3 | Suspendre le traitement. Reprendre l’administration du traitement lorsque les symptômes sont revenus au grade 0 ou 1. |
4 | Arrêter définitivement le traitement. | |
Récidive d’effets indésirables d’origine immunologique après leur retour à un grade de sévérité ≤ 1 (excepté pour la pneumopathie inflammatoire, voir ci-dessus) | 3 ou 4 | Arrêter définitivement le traitement. |
Autres effets indésirables | Grade de sévéritéa | Modification de traitement |
Réactions liées à la perfusion | 2 | Suspendre le traitement. Si la réaction disparaît dans l’heure qui suit l’arrêt de la perfusion, celle-ci pourra être reprise à 50 % du débit de perfusion initial ; sinon, recommencer la perfusion si les symptômes disparaissent avec une prémédication. Si les symptômes reviennent à un grade 2 avec une prémédication adéquate, arrêter définitivement le traitement. |
3 ou 4 | Arrêter définitivement le traitement. | |
a Les grades de toxicité correspondent à la classification du National Cancer Institute : Common Terminology Criteria for Adverse Events (CTCAE), version 5.0.
b ASAT = aspartate aminotransférase
c ALAT = alanine aminotransférase
d LSN = limite supérieure à la normale
e Chez les patientes présentant des métastases hépatiques et une augmentation du taux d’ASAT ou d’ALAT de grade 2 à l’initiation du traitement, si une augmentation des taux d’ASAT ou l’ALAT de
≥ 50 % par rapport à l’inclusion est observée pendant au moins 1 semaine, le traitement doit être arrêté.
f SJS = Syndrome de Stevens-Johnson
g NET = Nécrolyse épidermique Toxique (syndrome de Lyell)
h DRESS = Syndrome d’hypersensibilité médicamenteuse
Carte patiente
Tous les prescripteurs de JEMPERLI doivent informer les patientes de la « Carte Patiente », qui explique la démarche à suivre en cas de survenue de tout symptôme évocateur d’un effet indésirable d’origine immunologique. Le médecin doit remettre la « Carte Patiente » à chaque patiente.
Populations particulières
Personnes âgées
Aucune adaptation posologique n’est recommandée chez les patientes âgées de 65 ans et plus.
Les données cliniques sur l’utilisation de dostarlimab chez des patientes âgées de 75 ans et plus sont limitées (voir section 5.1).
Insuffisance rénale
Aucune adaptation posologique n’est recommandée chez les patientes présentant une insuffisance rénale légère ou modérée. Les données chez les patientes présentant une insuffisance rénale sévère ou une maladie rénale à un stade terminal et sous dialyse sont limitées (voir section 5.2).
Insuffisance hépatique
Aucune adaptation posologique n’est recommandée chez les patientes présentant une insuffisance hépatique légère. Les données chez les patientes présentant une insuffisance hépatique modérée sont limitées et aucune donnée n’est disponible chez les patientes présentant une insuffisance hépatique sévère (voir section 5.2).
Population pédiatrique
La sécurité et l’efficacité de JEMPERLI chez l’enfant et l’adolescente âgées de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
JEMPERLI doit être utilisé exclusivement en perfusion intraveineuse. JEMPERLI doit être administré en perfusion intraveineuse à l’aide d’une pompe à perfusion intraveineuse pendant 30 minutes.
JEMPERLI ne doit pas être administré par injection rapide ou en bolus intraveineux.
Pour les instructions sur la dilution du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables associés le plus fréquemment au dostarlimab sont d’origine immunologique. La plupart de ces effets indésirables, incluant des effets indésirables sévères, se sont résolus après l’initiation d’un traitement médical approprié ou l’arrêt de dostarlimab (voir « Description de certains effets indésirables »).
Dostarlimab en monothérapie
La sécurité du dostarlimab a été évaluée chez 605 patients atteintes d’un cancer de l’endomètre ou d’autres tumeurs solides avancées, qui ont reçu du dostarlimab en monothérapie dans le cadre de l’étude GARNET, incluant 153 patientes qui présentaient un cancer de l’endomètre dMMR/MSI-H avancé ou récidivant. Les patients ont reçu 500 mg de dostarlimab toutes les 3 semaines pour les 4 premiers cycles, puis 1 000 mg toutes les 6 semaines pour tous les cycles suivants.
Chez les patients qui présentaient des tumeurs solides avancées ou récidivantes (N = 605), les effets indésirables les plus fréquents (> 10%) étaient : anémie (28,6 %), diarrhées (26,0 %), nausées (25,8 %), vomissements (19,0 %), arthralgie (17,0 %), prurit (14,2 %), éruption cutanée (13,2%), fièvre (12,4%), augmentation de l'aspartate aminotransférase (11,2 %) et hypothyroïdie (11,2 %). JEMPERLI a été arrêté définitivement chez 38 patientes (6,3 %) en raison d’effets indésirables, la plupart d’origine immunologique. Les effets indésirables étaient graves chez 11,2 % des patients ; les effets indésirables les plus graves étaient les effets indésirables d’origine immunologiques (voir rubrique 4.4).
Le profil de sécurité des patientes atteintes d’un cancer de l’endomètre dMMR/MSI-H dans l’étude GARNET (N=153) n’était pas différent de celui de l’ensemble de la population sous monothérapie présenté dans le Tableau 4.
Dostarlimab en association avec le carboplatine et le paclitaxel
La sécurité du dostarlimab a été évaluée chez 241 patientes présentant un cancer de l’endomètre avancé ou récidivant qui ont reçu dostarlimab en association avec le carboplatine et le paclitaxel dans l’étude RUBY. Les patientes ont reçu des doses de 500 mg de dostarlimab toutes les 3 semaines pendant 6 cycles puis 1000 mg toutes les 6 semaines pour tous les cycles suivants.
Chez les patientes avec un cancer de l’endomètre avancé ou récurrent (N=241), les effets indésirables les plus fréquents (≥ 10%) étaient : éruption cutanée (23,2%), éruption cutanée maculopapuleuse (14,5%), hypothyroïdie (14,5%), fièvre (12,9%) alanine aminotransférase augmentée (12,9%), aspartate aminotransférase augmentée (12,0%), et sècheresse cutanée (10,0%). JEMPERLI a été arrêté définitivement chez 12 patientes (5,0%) en raison d’effets indésirables ; la plupart d’origine immunologique. Les effets indésirables étaient graves chez 5,8% des patientes. L’effet indésirable grave le plus fréquent (>1 %) était la fièvre (2,9 %). L’effet indésirable d’origine immunologique le plus fréquent (>10 %) était l'hypothyroïdie (12,0 %). L’effet indésirable d’origine immunologique le plus fréquent (>1 %) entraînant l’arrêt du traitement était l’éruption maculo-papuleuse (1,2 %) (voir rubrique 4.4).
Liste tabulée des effets indésirables
La liste des effets indésirables rapportés dans les essais cliniques avec le dostarlimab en monothérapie ou en combinaison avec la chimiothérapie sont répertoriés dans le tableau 4 par classe de systèmes d’organes et par fréquence. Les fréquences des effets indésirables listés dans la colonne « dostarlimab en monothérapie » sont basées sur la fréquence des événements indésirables toutes causes confondues identifiés chez 605 patients atteints de tumeurs solides avancées ou récidivantes issus de l’étude GARNET exposés au dostarlimab en monothérapie pour une durée médiane de traitement de 24 semaines (extrêmes : 1 semaine à 229 semaines). Sauf indication contraire, les fréquences des effets indésirables listées dans la colonne « dostarlimab en association avec la chimiothérapie » sont basées sur la fréquence d’évènements indésirables toutes causes confondues identifiées chez 241 patientes atteintes d’un cancer de l’endomètre avancé ou récidivant provenant de l’étude RUBY exposées au dostarlimab en association avec le carboplatine et le paclitaxel pour une durée médiane de traitement de 43 semaines (intervalle allant de 3 à 193 semaines). Pour des informations supplémentaires de sécurité lorsque le dostarlimab est administré en association avec le carboplatine et le paclitaxel, veuillez-vous référer aux informations de prescription respectives des produits administrés en association.
Les effets indésirables connus pour survenir avec le dostarlimab en monothérapie ou avec le carboplatine ou le paclitaxel donnés seuls peuvent survenir pendant le traitement avec ces médicaments en association, même si ces réactions n’étaient pas rapportées dans les études cliniques avec le dostarlimab en association avec le carboplatine et le paclitaxel.
Ces effets sont présentés par classe de systèmes d’organes et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) de fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 4 : Effets indésirables chez les patients traités par dostarlimab
| Dostarlimab en monothérapie | Dostarlimab en association avec la chimiothérapie |
Affections hématologiques et du système lymphatique | ||
Très fréquent | Anémiea |
|
Affections endocriniennes | ||
Très fréquent | Hypothyroïdie* b | Hypothyroïdiee |
Fréquent | Hyperthyroïdie*, insuffisance surrénalienne* | Hyperthyroïdie |
Peu fréquent | Thyroïdite*c, hypophysited | Thyroïdite, insuffisance surrénalienne |
Troubles du métabolisme et de la nutrition | ||
Peu fréquent | Diabète sucré de type I, acidocétose diabétique | Diabète sucré de type I |
Troubles du système nerveux | ||
Peu fréquent | Encéphalite, myasthénie grave | Syndrome myasthénique†, |
Affections oculaires | ||
Peu fréquent | Uvéiteg | Uvéite |
Affections cardiaques | ||
Peu fréquent |
| Myocardite†h |
Affections respiratoires, thoraciques et médiastinales | ||
Fréquent | Pneumopathie | Pneumopathie inflammatoire |
Affections gastro-intestinales | ||
Très fréquent | Diarrhées, nausées, |
|
Fréquent | Colite*j,pancréatitek, gastrite | Colite†l, pancréatite |
Peu fréquent | Œsophagite | Gastrite à médiation immunitaire† , vascularite gastro-intestinale† |
Affections hépatobiliaires | ||
Fréquent | Hépatite*m |
|
Affections de la peau et du tissu sous-cutané | ||
Très fréquent | Eruption cutanée*n, prurit | Eruption cutanéeo, sècheresse cutanée |
Affections musculosquelettiques et systémiques | ||
Très fréquent | Arthralgie* |
|
Fréquent | Myalgie |
|
Peu fréquent | Arthrite à médiation immunitaire, pseudopolyarthrite rhizomélique, myosite à médiation immunitaire | Arthrite à médiation immunitaire, myosite† |
Troubles du rein et des voies urinaires | ||
Peu fréquent | Néphrite*p |
|
Troubles généraux et anomalies au site d’administration | ||
Très fréquent | Fièvre | Fièvre |
Fréquent | Frissons |
|
Peu fréquent |
| Syndrome de réponse inflammatoire systémique† |
Investigations | ||
Très fréquent | Augmentation des transaminasesq | Alanine aminotransférase augmentée, |
Lésions, intoxications et complications liées aux procédures | ||
Fréquent | Réaction à la perfusion*r |
|
†Inclut les événements identifiés dans d'autres essais cliniques chez des patients atteints de tumeurs solides et recevant le dostarlimab en association avec différents types de thérapies anticancéreuses.
*Voir la rubrique « Description de certains effets indésirables »
aInclut anémie et anémie hémolytique auto-immune
bInclut hypothyroïdie et hypothyroïdie auto-immune
cInclut thyroïdite et thyroïdite auto-immune
d Inclut hypophysite et hypophysite lymphocytaire
eInclut hypothyroïdie et hypothyroïdie à médiation immunitaire
f Inclut le syndrome de Guillain-Barré et la polyneuropathie démyélinisante
gInclut uvéite et iridocyclite
hInclut myocardite et myocardite à médiation immunitaire
iInclut pneumopathie inflammatoire, pneumopathie interstitielle diffuse et maladie pulmonaire à médiation immunitaire
jInclut colite, entérocolite et entérocolite à médiation immunitaire
kInclut pancréatite et pancréatite aigue
lInclut colite et entérite
mInclut hépatite, hépatite auto-immune et cytolyse hépatique
nInclut éruption cutanée, éruption cutanée maculo-papuleuse, érythème, éruption cutanée maculaire, éruption cutanée prurigineuse, éruption cutanée érythémateuse, éruption cutanée papuleuse, érythème polymorphe, toxicité cutanée, éruption cutanée d’origine médicamenteuse, éruption cutanée toxique, éruption cutanée exfoliative et pemphigoïde
oInclut éruption cutanée et éruption cutanée maculo-papuleuse
pInclut néphrite et néphrite tubulo-interstitielle
q.Inclut augmentation des transaminases, alanine aminotransférase augmentée, aspartate aminotransférase augmentée et hypertransaminasémie
r Inclut réactions liées à la perfusion et hypersensibilité.
Description de certains effets indésirables
Les effets indésirables décrits ci-dessous sont basés sur les données enregistrées dans une base de données de sécurité combinée de 605 patients de l’étude GARNET recevant dostarlimab pour un cancer de l’endomètre ou d’autres tumeurs solides avancées. Les effets indésirables d’origine immunologique ont été définis sur la base des événements de grade 2 et au-delà ; les fréquences indiquées ci-dessous excluent les événements de grade 1. Les recommandations de prise en charge de ces effets indésirables sont décrites à la rubrique 4.2.
Effets indésirables d’origine immunologique (voir rubrique 4.4)
Pneumopathie inflammatoire d’origine immunologique
Une pneumopathie inflammatoire d’origine immunologique est survenue chez 14 (2,3 %) patients recevant dostarlimab, dont des cas de grade 2 (1,3 %), 3 (0,8 %) ou 4 (0,2 %). Une pneumopathie inflammatoire a entraîné l’arrêt du dostarlimab chez 8 patients (1,3 %).
Des corticoïdes systémiques (prednisone ≥ 40 mg/jour ou équivalent) ont été nécessaires chez 11(78,6 %) patients qui ont présenté une pneumopathie inflammatoire. La pneumopathie inflammatoire s’est résolue chez 11 patients (78,6 %).
Colite d’origine immunologique
Une colite est survenue chez 8 patients (1,3 %), dont des cas de grade 2 (0,7 %) ou 3 (0,7 %). Une colite n’a pas entraîné l’arrêt de dostarlimab chez les patients.
Des corticoïdes systémiques (prednisone ≥ 40 mg/jour ou équivalent) ont été nécessaires chez 5 patients (62,5 %). La colite s’est résolue chez 5 patients (62,5 %) ayant eu une colite.
Hépatite d’origine immunologique
Une hépatite d’une sévérité de grade 3 est survenue chez 3 patients (0,5 %). Des corticoïdes systémiques (prednisone ≥ 40 mg/jour ou équivalent) ont été nécessaires chez 2 patients (66,7 %). L’hépatite a entraîné l’arrêt du dostarlimab chez 1 patient (0,2 %) et s’est résolue chez 2 patients sur 3.
Endocrinopathies à médiation immunitaire
Une hypothyroïdie est survenue chez 46 patients (7,6 %), toutes de grade 2. L’hypothyroïdie n’a pas entraîné l’arrêt du dostarlimab et s’est résolue chez 17 patients (37,0 %).
Une hyperthyroïdie est survenue chez 14 patients (2,3 %), dont des cas de grade 2 ou 3 chez 2,1 % et 0,2 % de patients respectivement. L’hyperthyroïdie n’a pas entraîné l’arrêt du dostarlimab et s’est résolue chez 10 patients (71,4 %).
Une thyroïdite est survenue chez 3 patients (0,5 %), toutes étaient des cas de grade 2. La thyroïdite ne s’est pas résolue chez ces patients mais n’a pas nécessité l’arrêt du dostarlimab.
Une insuffisance surrénalienne est survenue chez 7 patients (1,2 %), dont des cas de grade 2 ou 3 chez 0,5 % et 0,7 % de patients respectivement. L’insuffisance surrénalienne a entraîné l’arrêt du dostarlimab chez 1 patient (0,2 %) et s’est résolue chez 4 patients (57,1 %).
Néphrite à médiation immunitaire
Une néphrite, incluant aussi une néphrite tubulo-intersticielle, est survenue chez 3 patients (0,5 %) ; toutes de grade 2. Des corticoïdes systémiques (prednisone ≥ 40 mg/jour ou équivalent) ont été nécessaires chez 2 patients (66,7 %) ayant présentés une néphrite. La néphrite a entraîné l’arrêt du dostarlimab chez 1 patient (0,2%) et s’est résolue chez les trois patients.
Eruption cutanée d’origine immunologique
Une éruption cutanée d’origine immunologique (éruption cutanée, éruption cutanée maculo-papuleuse, éruption cutanée maculaire, éruption cutanée prurigineuse, pemphigoïde, éruption cutanée d’origine médicamenteuse, toxicité cutanée, éruption cutanée toxique) est survenue chez 31 patients (5,1 %), dont des cas de grade 3 chez 9 patients (1,5 %) recevant dostarlimab. Le temps médian de survenue de l’éruption cutanée était de 57 jours (extrêmes de 2 jours à 1485 jours). Des corticoïdes systémiques (prednisone ≥ 40 mg/jour ou équivalent) ont été nécessaires chez 9 patients (29 %) ayant présentés une éruption cutanée. L’éruption cutanée a nécessité l’arrêt du dostarlimab chez un patient (0,2%) et s’est résolue chez 24 patients (77,4 %).
Arthralgie d’origine immunologique
Une arthralgie d’origine immunologique est survenue chez 34 patients (5,6 %). Une arthralgie d’origine immunologique de grade 3 a été rapporté chez 5 patients (0,8 %) recevant du dostarlimab. Le temps médian de survenue de l’arthralgie était de 94,5 jours (extrêmes de 1 jour à 840 jours). Des corticoïdes systémiques (prednisone≥ 40 mg/jour ou équivalent) ont été nécessaires chez 3 patients (8,8 %) ayant présenté une arthralgie. L’arthralgie a nécessité l’arrêt du dostarlimab chez un patient (0,2%) et s’est résolue chez 19 patients (55,9 %) ayant présenté une arthralgie.
Réactions liées à la perfusion
Des réactions liées à la perfusion, parmi lesquelles des réactions d’hypersensibilité, ont été observées chez 6 patients (1,0 %), dont 0,3 % de grade 2 et 0,2 % de grade 3. Tous ces patients ont guéri de leur réaction à la perfusion.
Effets de la classe des inhibiteurs de point de contrôle immunitaire
Des cas avec l’effet indésirable suivant ont été rapportés au cours du traitement par d’autres inhibiteurs de point de contrôle immunitaire, qui peuvent également survenir au cours du traitement par dostarlimab : maladie cœliaque ; insuffisance pancréatique exocrine.
Immunogénicité
Dans l’étude GARNET, les anticorps anti-médicaments ont été dosés chez 315 patients ayant reçu dostarlimab ; l’incidence des anticorps anti-médicaments apparus sous traitement par dostarlimab était de 2,5 %. Des anticorps neutralisants ont été détectés chez 1,3 % des patientes. La co-administration avec le carboplatine et le paclitaxel n’a pas affecté l’immunogénicité du dostarlimab. Dans l’étude RUBY, parmi les 225 patientes qui étaient traitées par le dostarlimab en association avec le carboplatine et le paclitaxel et dont la présence d’anticorps anti-médicaments était évaluable, il n’y a eu aucune incidence d’anticorps anti-médicaments apparus sous traitement par dostarlimab ou d’anticorps neutralisants apparus sous traitement.
Chez les patientes ayant développé des anticorps anti-dostarlimab, aucune altération de l’efficacité ou de la sécurité du dostarlimab n’a été mise en évidence.
Population âgée
Sur les 605 patientes traitées en monothérapie avec dostarlimab, 51,6% avaient moins de 65 ans, 36,9 % avaient entre 65 et moins de 75 ans et 11,5 % avaient 75 ans ou plus. Sur les 241 patientes traitées par dostarlimab en association avec carboplatine-paclitaxel, 52,3 % avaient moins de 65 ans, 36,5 % avaient entre 65 et moins de 75 ans, et 11,2 % avaient 75 ans ou plus. Globalement, aucune différence du point de vue de la tolérance n’a été signalée entre les patientes âgées (≥ 65 ans) et jeunes (< 65 ans).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique | Luxembourg |
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
GlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Irlande
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/21/1538/001
10. DATE DE MISE À JOUR DU TEXTE
25/04/2025 (V10)
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4309209 | JEMPERLI 500 mg solution à diluer pour perfusion | - | € 5631,07 | Oui | - | - |