RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Cotellic 20 mg, comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient de l’hémifumarate de cobimetinib équivalent à 20 mg de cobimetinib.
Excipient(s) à effet notoire
Chaque comprimé pelliculé contient 36 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé.
Comprimé pelliculé rond et blanc d’environ 6,6 mm de diamètre, gravé « COB » sur une face.
4. DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Cotellic est indiqué en association au vemurafenib dans le traitement des patients adultes atteints d’un mélanome non résécable ou métastatique porteur d’une mutation BRAF V600 (voir rubriques 4.4 et 5.1).
4.2 Posologie et mode d’administration
Le traitement par Cotellic en association au vemurafenib doit être initié et supervisé par un médecin qualifié expérimenté dans l’utilisation des traitements anticancéreux.
Avant le début de ce traitement, la présence de la mutation BRAF V600 doit être confirmée par un test validé (voir rubriques 4.4 et 5.1).
Posologie
La dose recommandée de Cotellic est de 60 mg (soit 3 comprimés à 20 mg) une fois par jour.
La prise de Cotellic suit un cycle de 28 jours. Chaque dose se compose de trois comprimés de 20 mg (soit 60 mg) et doit être prise une fois par jour pendant 21 jours consécutifs (jours 1 à 21- période de traitement), suivis d’une période sans traitement de 7 jours (jours 22 à 28 – pause du traitement). Le cycle suivant de traitement par Cotellic doit commencer une fois que la période sans traitement de 7 jours s’est écoulée.
Pour toute information sur la posologie du vemurafenib, se référer au Résumé des Caractéristiques du Produit (RCP).
Durée du traitement
Le traitement par Cotellic doit être poursuivi tant que le patient en tire un bénéfice ou jusqu’à la survenue d’une toxicité inacceptable (voir le Tableau 1 ci-dessous).
Omission d’une dose
Si une dose est omise, elle peut être prise jusqu’à 12 heures avant la dose suivante afin de maintenir la fréquence d’administration à une prise par jour.
Vomissement
En cas de vomissement suite à l’administration de Cotellic, le patient ne doit pas prendre de dose supplémentaire le même jour, le traitement doit être poursuivi le lendemain de la façon prescrite.
Adaptations posologiques générales
La décision de réduire la dose de l’un ou l’autre des médicaments ou des deux doit reposer sur l’évaluation par le prescripteur de la sécurité ou de la tolérance de chaque patient. L’adaptation posologique de Cotellic est indépendante de celle du vemurafenib.
Si des doses n’ont pas été prises suite à une toxicité, celles-ci ne doivent pas être remplacées. Lorsque la dose a été réduite, elle ne doit pas être augmentée ultérieurement.
Le Tableau 1 ci-dessous présente les recommandations générales d’adaptation posologique de Cotellic.
Tableau 1 Recommandations générales d’adaptations posologiques de Cotellic
Grade (CTC-AE) * | Dose recommandée de Cotellic |
Grade 1 ou Grade 2 (tolérable) | Aucune réduction de dose. Maintenir Cotellic à la dose de 60 mg une fois par jour (3 comprimés) |
Grade 2 (intolérable) |
|
1re survenue | Interrompre le traitement jusqu’à un grade ≤ 1, reprendre le traitement à la dose de 40 mg une fois par jour (2 comprimés) |
2e survenue | Interrompre le traitement jusqu’à un grade ≤ 1, reprendre le traitement à la dose de 20 mg une fois par jour (1 comprimé) |
3e survenue | Envisager l’arrêt définitif du traitement |
* L’intensité des événements indésirables cliniques est évaluée par les Critères terminologiques communs pour les événements indésirables v4.0 (CTC-AE)
Recommandations d’adaptations posologiques en cas d’hémorragie
Evénements de grade 4 ou hémorragie cérébrale : le traitement par Cotellic doit être interrompu. En cas d’événement hémorragique attribué à Cotellic, le traitement par Cotellic doit être arrêté définitivement.
Evénements de grade 3 : le traitement par Cotellic doit être interrompu pendant son évaluation afin d'éviter toute aggravation potentielle de l'événement. Aucune donnée n’est disponible sur l’efficacité des adaptations posologiques de Cotellic en cas d’événements hémorragiques. La reprise du traitement par Cotellic doit se baser sur une évaluation clinique. Le traitement par vemurafenib peut être poursuivi, si indiqué, en cas d’interruption du traitement par Cotellic.
Recommandations d’adaptations posologiques en cas de dysfonction ventriculaire gauche
En cas de symptômes cardiaques attribués à Cotellic et qui ne s’améliorent pas après une interruption temporaire, un arrêt définitif du traitement par Cotellic doit être envisagé.
Tableau 2 : Recommandations d’adaptations posologiques de Cotellic chez les patients présentant une diminution de la fraction d’éjection ventriculaire gauche (FEVG) par rapport aux valeurs initiales
Patient | Valeur de la FEVG | Adaptation posologique recommandée de Cotellic | Valeur de la FEVG après une interruption du traitement | Dose quotidienne recommandée de Cotellic |
Asymptomatique | ≥ 50 % | Poursuivre le traitement à la même posologie | N/A | N/A |
< 40 % | Interrompre le traitement pendant 2 semaines | Diminution absolue par rapport aux valeurs initiales <10 % | 1re survenue: 40 mg | |
2e survenue : 20 mg | ||||
3e survenue : | ||||
< 40 % | Arrêt permanent | |||
Symptomatique | N/A | Interrompre le traitement pendant 4 semaines | Asymptomatique et diminution absolue par rapport aux valeurs initiales <10 % | 1re survenue : 40 mg |
2e survenue : 20 mg | ||||
3e survenue : | ||||
Asymptomatique et < 40 % (ou diminution absolue par rapport aux valeurs initiales ≥10 %) | Arrêt permanent | |||
Symptomatique quelle que soit la valeur de la FEVG | Arrêt permanent |
N/A = Non applicable
En cas d’adaptation posologique de Cotellic, le traitement par le vemurafenib peut être poursuivi (si cliniquement indiqué).
Recommandations d’adaptations posologiques en cas de rhabdomyolyse et d’élévations de la créatine phosphokinase (CPK)
Rhabdomyolyse ou élévations symptomatiques de la CPK:
Le traitement par Cotellic doit être interrompu. Si la rhabdomyolyse ou l’élévation symptomatique de la CPK ne s’améliore pas dans les 4 semaines suivant l’interruption, le traitement par Cotellic doit être définitivement arrêté. Si la sévérité s’améliore d'au moins un grade dans les 4 semaines, le traitement par Cotellic peut être repris, si cliniquement indiqué, à une dose réduite de 20 mg. Les patients doivent être étroitement surveillés.
Le traitement par vemurafenib peut être poursuivi lors de toute modification de l’administration de Cotellic.
Elévations asymptomatiques de la CPK:
Grade 4: le traitement par Cotellic doit être interrompu. Si l'élévation de la CPK ne s'améliore pas à un grade ≤ 3 dans les 4 semaines suivant l'interruption, le traitement par Cotellic doit être définitivement arrêté. Si l’élévation s’améliore à un grade ≤ 3 dans les 4 semaines, Cotellic peut être repris, si cliniquement indiqué, à une dose réduite de 20 mg et les patients doivent être étroitement surveillés.
Le traitement par vemurafenib peut être poursuivi lors de toute modification de l’administration de Cotellic.
Grade ≤ 3: si une rhabdomyolyse est écartée, aucune adaptation posologique de Cotellic n’est nécessaire.
Recommandations d’adaptations posologiques de Cotellic utilisé en association avec le vemurafenib
Anomalies du bilan hépatique
En cas d’anomalie du bilan hépatique de grade 1 et 2, Cotellic et le vemurafenib doivent être poursuivis à la dose prescrite.
Grade 3 : Cotellic doit être maintenu à la dose prescrite. La dose de vemurafenib peut être réduite si cliniquement approprié. Se référer au RCP de vemurafenib.
Grade 4 : Le traitement par Cotellic et vemurafenib doit être interrompu. Si les anomalies du bilan hépatique s’améliorent à un grade ≤ 1 dans les 4 semaines, Cotellic peut être réinstauré à une dose réduite de 20 mg et le vemurafenib à la dose cliniquement appropriée, conformément à son RCP.
Si les anomalies du bilan hépatique ne s’améliorent pas à un grade ≤ 1 dans les 4 semaines ou si des anomalies du bilan hépatique de grade 4 réapparaissent après une amélioration initiale, le traitement par Cotellic et vemurafenib doit être arrêté.
Photosensibilité
Une photosensibilité de grade ≤ 2 (tolérable) doit être prise en charge par un traitement symptomatique.
En cas de photosensibilité de grade 2 (intolérable) ou de grade ≥ 3, Cotellic et le vemurafenib doivent être interrompus jusqu’à la résolution à un grade ≤ 1. Le traitement peut être réinstauré sans modification de la dose de Cotellic. La dose de vemurafenib doit être réduite si cela est cliniquement indiqué ; pour plus d’information se référer à son RCP.
Éruption cutanée
Des éruptions cutanées peuvent survenir avec Cotellic ou le vemurafenib. La dose de Cotellic et/ou de vemurafenib peut être soit interrompue temporairement soit réduite selon les manifestations cliniques.
En outre :
Une éruption cutanée de grade ≤ 2 (tolérable) doit être prise en charge par un traitement symptomatique. L’administration de Cotellic peut être poursuivie sans adaptation.
En cas d’éruption cutanée acnéiforme de grade 2 (intolérable) ou grade ≥ 3 : les recommandations générales d’adaptations posologiques de Cotellic du Tableau 1 doivent être suivies. L’administration du vemurafenib peut être poursuivie lorsque le traitement par Cotellic est modifié (si cliniquement indiqué).
En cas d’éruption cutanée non acnéiforme ou maculo-papuleuse de grade 2 (intolérable) ou grade ≥ 3 : l’administration de Cotellic peut être poursuivie sans adaptation si cliniquement indiquée. L’administration du vemurafenib peut être suspendue temporairement et/ou poursuivie à une dose réduite ; pour plus d’information se référer à son RCP.
Allongement de l’intervalle QT
Si l’intervalle QTc dépasse 500 ms au cours du traitement, se référer au RCP du vemurafenib (voir rubrique 4.2) pour les mesures d’adaptations posologiques de vemurafenib. Aucune adaptation posologique de Cotellic n’est nécessaire lorsqu’il est pris en association au vemurafenib.
Populations particulières
Patients âgés
Aucune adaptation posologique de Cotellic n’est nécessaire chez les patients âgés de 65 ans et plus.
Insuffisants rénaux
Aucune adaptation posologique n’est recommandée chez les patients présentant une insuffisance rénale légère ou modérée, sur la base de l’analyse pharmacocinétique de population (voir rubrique 5.2). Les données chez les patients présentant une insuffisance rénale sévère étant limitées, un effet ne peut être exclu. Cotellic doit être utilisé avec prudence chez les patients présentant une insuffisance rénale modérée à sévère.
Insuffisants hépatiques
Aucune adaptation posologique n’est recommandée chez les patients présentant une insuffisance hépatique. Les patients présentant une insuffisance hépatique sévère peuvent avoir des concentrations plasmatiques en cobimetinib libre augmentées par rapport aux patients ayant une fonction hépatique normale (voir rubrique 5.2). Des anomalies du bilan hépatique peuvent apparaître avec Cotellic, la prudence est de rigueur chez les patients présentant une insuffisance hépatique quel qu’en soit le degré (voir rubrique 4.4).
Patients non caucasiens
La sécurité et l’efficacité de Cotellic n’ont pas été établies chez des patients non caucasiens.
Population pédiatrique
La sécurité et l’efficacité de Cotellic n’ont pas été établies chez les enfants et les adolescents (âgés de moins de 18 ans). Les données actuellement disponibles sont décrites dans les rubriques 4.8, 5.1 et 5.2, mais aucune recommandation sur la posologie ne peut être faite.
Mode d’administration
Voie orale. Les comprimés de Cotellic doivent être avalés entiers avec de l’eau. Ils peuvent être pris avec ou sans aliments.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de tolérance
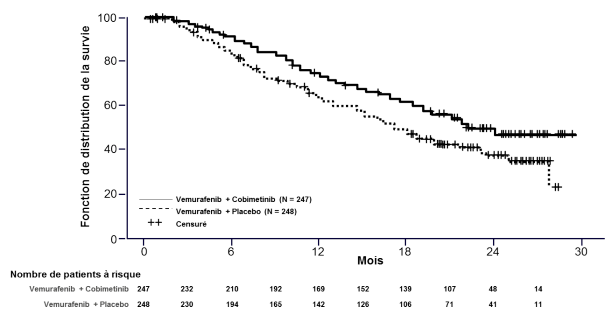
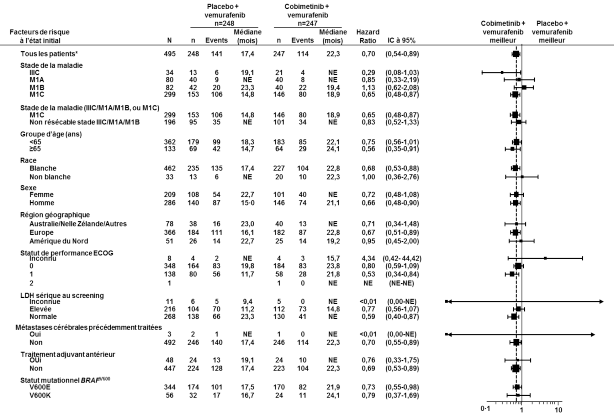
La sécurité de Cotellic en association au vemurafenib a été évaluée chez 247 patients présentant un mélanome avancé porteur d’une mutation BRAF V600 dans l’étude de phase III GO28141. Le délai médian de survenue des premiers événements indésirables de grade ≥ 3 était de 0,6 mois dans le bras Cotellic associé au vemurafenib versus 0,8 mois dans le bras placebo plus vemurafenib.
La sécurité de Cotellic en association au vemurafenib a également été évaluée chez 129 patients présentant un mélanome avancé porteur d’une mutation de BRAF V600 dans l’étude NO25395. Le profil de sécurité observé dans l’étude NO25395 était conforme à celui de l’étude GO28141.
Dans l’étude GO28141, les effets indésirables les plus fréquents (> 20 %) observés à une fréquence plus élevée dans le bras Cotellic associé au vemurafenib ont été diarrhée, éruption cutanée, nausée, pyrexie, réaction de photosensibilité, élévation de l’alanine aminotransférase, élévation de l’aspartate aminotransférase, élévation de la créatine phosphokinase sanguine et vomissements. Les effets indésirables les plus fréquents (> 20 %) observés à une fréquence plus élevée dans le bras placebo associé au vemurafenib ont été arthralgie, alopécie et hyperkératose. La fatigue a été observée à des fréquences similaires dans les deux bras.
Se reporter au RCP du vemurafenib pour la description complète de tous les effets indésirables liés à un traitement par vemurafenib.
Liste tabulée des effets indésirables
Les effets indésirables sont basés sur les résultats d’une étude de phase III (GO28141), multicentrique, randomisée, menée en double aveugle contre placebo, qui a évalué Cotellic en association avec le vemurafenib comparé au vemurafenib seul chez des patients présentant un mélanome non résécable (stade III) ou métastatique (stade IV) porteur d’une mutation BRAF V600 naïfs de tout traitement.
La fréquence des effets indésirables est basée sur l'analyse de la sécurité des patients traités par cobimetinib associé au vemurafenib avec une durée médiane de suivi de 11,2 mois (date de cut-off des données au 19 septembre 2014).
Les effets indésirables rapportés chez des patients atteints d’un mélanome sont listés ci-dessous par système organe-classe MedDRA, fréquence et grade de sévérité. La convention suivante a été utilisée pour la classification des fréquences :
Très fréquent ≥ 1/10
Fréquent ≥ 1/100 à < 1/10
Peu fréquent ≥ 1/1 000 à < 1/100
Rare ≥ 1/10 000 à 1/1 000
Très rare < 1/10 000
Le Tableau 3 liste les effets indésirables considérés reliés à l’utilisation de Cotellic. Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante et ont été rapportés selon les critères communs de toxicité NCI-CTCAE v 4.0 (common toxicity criteria) pour l’évaluation de la toxicité dans l’étude GO28141.
Tableau 3 : Effets indésirables survenus chez des patients traités par Cotellic en association au vemurafenib dans l’étude GO28141^
Système | Très fréquent | Fréquent | Peu fréquent |
Tumeurs bénignes, malignes et non précisées (dont kystes et polypes) |
| Carcinome basocellulaire, Carcinome épidermoïde cutané**, Kératoacanthome** |
|
Affections hématologiques et du système lymphatique | Anémie |
|
|
Troubles du métabolisme et de la nutrition |
| Déshydratation, Hypophosphatémie, Hyponatrémie, Hyperglycémie |
|
Affections oculaires | Rétinopathie séreusea, Vision trouble | Déficience visuelle |
|
Affections vasculaires | Hypertension, Hémorragie* |
|
|
Affections respiratoires, thoraciques et médiastinales |
| Pneumopathie inflammatoire |
|
Affections gastro-intestinales | Diarrhée, Nausées, Vomissements, Stomatite |
|
|
Affections de la peau et du tissus sous-cutané | Photosensibilitéb, Eruption cutanée, Eruption cutanée maculo-papuleuse, Dermatite acnéiforme, Hyperkératose**, Pruritc, Sécheresse cutanéec |
|
|
Affections musculo-squelettiques et systémiques |
|
| Rhabdomyolyse*** |
Troubles généraux et anomalies au site d'administration | Pyrexie, Frissons, Oedème périphériquec |
|
|
Investigations | Elévation de la CPK sanguine, Elévation des ALAT, Elévation des ASAT, Elévation des Gamma-Glutamyltransferases (GGT), Elévation des phosphatases alcalines sanguines | Diminution de la fraction d’éjection, Elévation de la bilirubine sanguine |
|
^ date de cut-off des données au 19 septembre 2014
* se reporter au paragraphe Hémorragie à la rubrique « Description des effets indésirables sélectionnés »
** se reporter au paragraphe Carcinome épidermoïde cutané, kératoacanthome et hyperkératose
à la rubrique « Description des effets indésirables sélectionnés »
*** se reporter au paragraphe Rhabdomyolyse à la rubrique « Description des effets indésirables sélectionnés »
a incluant les évènements de choriorétinopathie et décollement de la rétine révélateurs d’une rétinopathie séreuse (voir rubrique 4.4)
b valeur regroupant les cas rapportés de réaction de photosensibilité, coups de soleil, dermatite solaire, élastose actinique
c effets indésirables identifiés dans une étude avec le cobimetinib en monothérapie (ML29733 ; étude américaine). Cependant, ces effets indésirables ont également été rapportés avec l'association cobimetinib plus vemurafenib dans des essais cliniques menés chez des patients atteints de mélanome non résécable ou métastatique.
Description des effets indésirables sélectionnés
Hémorragie
Des événements hémorragiques ont été rapportés plus fréquemment dans le bras Cotellic associé au vemurafenib que dans le bras placebo associé au vemurafenib (tous types et tous grades : 13 % versus 7 %). Le délai médian de première survenue était de 6,1 mois dans le bras Cotellic associé au vemurafenib.
La majorité des événements ont été de grade 1 ou 2, non graves. La plupart des événements se sont résolus sans adaptation de dose de Cotellic. Des événements hémorragiques majeurs (y compris hémorragies intracrâniennes et gastro-intestinales) ont été rapportés depuis la commercialisation. Le risque d'hémorragie peut être augmenté avec l'utilisation concomitante d'un traitement antiplaquettaire ou anticoagulant. En cas d'hémorragie, le traitement doit être adapté selon le tableau clinique (voir rubriques 4.2 et 4.4).
Rhabdomyolyse
Des cas de rhabdomyolyse ont été rapportés depuis la commercialisation. Des signes ou symptômes de rhabdomyolyse justifient une évaluation clinique et un traitement adaptés, ainsi qu’une adaptation de dose ou l’arrêt de Cotellic selon la sévérité de l'effet indésirable (voir rubriques 4.2 et 4.4).
Photosensibilité
Une photosensibilité a été observée avec une fréquence supérieure dans le bras Cotellic associé au vemurafenib par rapport au bras placebo associé au vemurafenib (47 % versus 35 %). La majorité des événements était de grade 1 ou 2, des événements de grade ≥ 3 étant survenus chez 4 % des patients du bras Cotellic associé au vemurafenib versus 0 % dans le bras placebo associé au vemurafenib.
Il n’a été observé aucune tendance en termes de délai de survenue d’événements de grade ≥ 3. Les événements à type de photosensibilité de grade ≥ 3 dans le bras Cotellic associé au vemurafenib ont été traités par des médicaments topiques et une interruption temporaire du traitement par cobimetinib et vemurafenib (voir rubrique 4.2).
Aucun signe de phototoxicité n’a été observé avec Cotellic administré en monothérapie.
Carcinome épidermoïde cutané, kératoacanthome et hyperkératose
Des carcinomes épidermoïdes cutanés ont été rapportés avec une fréquence inférieure dans le bras Cotellic associé au vemurafenib par rapport au bras placebo associé au vemurafenib (tous les grades : 3 % versus 13 %). Des kératoacanthomes ont été observés avec une fréquence inférieure dans le bras Cotellic associé au vemurafenib par rapport au bras placebo associé au vemurafenib (tous grades : 2 % versus 9 %). Une hyperkératose a été rapportée avec une fréquence inférieure dans le bras Cotellic associé au vemurafenib par rapport au bras placebo associé au vemurafenib (tous grades : 11 % versus 30 %).
Rétinopathie séreuse
Des rétinopathies séreuses ont été rapportées chez des patients traités par Cotellic (voir rubrique 4.4). Un examen ophtalmologique est recommandé chez les patients signalant l’apparition de troubles visuels ou l’aggravation de troubles visuels existants. Une rétinopathie séreuse peut être prise en charge par une interruption du traitement, une réduction de dose ou par l’arrêt du traitement (voir le Tableau 1 à la rubrique 4.2).
Dysfonction ventriculaire gauche
Une diminution de la FEVG par rapport aux valeurs initiales a été observée chez des patients traités par Cotellic (voir rubrique 4.4). La FEVG doit être contrôlée avant le début du traitement afin d’évaluer les valeurs initiales, après le premier mois de traitement et tous les trois mois par la suite, ou selon les indications cliniques jusqu’à l’arrêt du traitement. La diminution de la FEVG par rapport aux valeurs initiales peut être prise en charge par une suspension temporaire du traitement, une réduction posologique ou un arrêt du traitement (voir rubrique 4.2).
Anomalies biologiques
Anomalie du bilan hépatique
Des anomalies du bilan hépatique, telles que des augmentations des ALAT, ASAT et PAL ont été observées chez les patients traités par Cotellic associé au vemurafenib (voir rubrique 4.4). Des contrôles du bilan hépatique doivent être réalisés avant l’initiation du traitement en association et tous les mois pendant le traitement, ou plus fréquemment si cliniquement indiqué (voir rubrique 4.2).
Elévation de la créatine phosphokinase sanguine
Des cas d’élévation asymptomatique du taux de CPK dans le sang ont été observés à une fréquence plus élevée dans le bras Cotellic associé au vemurafenib comparé au bras placebo associé au vemurafenib dans l’étude GO28141 (voir rubriques 4.2 et 4.4). Un évènement de rhabdomyolyse a été observé dans chaque bras de traitement de l’étude avec des hausses simultanées de CPK dans le sang.
Le Tableau 4 précise la fréquence des anomalies mesurées du bilan hépatique et de l’élévation de la créatine phosphokinase de tous grades et de grades 3-4.
Tableau 4 : Exploration fonctionnelle du foie et autres analyses de laboratoire issues de l’étude de phase III GO28141
Changements dans les analyses de laboratoire rapportées | Cobimetinib plus vemurafenib | Placebo plus | ||
| Tous Grades | Grades 3‑4 | Tous Grades | Grades 3‑4 |
Exploration fonctionnelle du foie | ||||
Elévation de la PAL | 69 | 7 | 55 | 3 |
Elévation des ALAT | 67 | 11 | 54 | 5 |
Elévation des ASAT | 71 | 7 | 43 | 2 |
Elévation des GGT | 62 | 20 | 59 | 17 |
Elévation de la bilirubine sanguine | 33 | 2 | 43 | 1 |
Autres anomalies biologiques | ||||
Elévation de la CPK sanguine | 70 | 12 | 14 | <1 |
Populations particulières
Patients âgés
Dans l'étude de phase III menée avec Cotellic en association avec le vemurafenib chez les patients atteints d’un mélanome non résécable ou métastatique (n = 247), 183 patients (74 %) avaient moins de 65 ans, et 44 patients (18 %) avaient entre 65 et 74 ans, 16 (6 %) avaient entre 75 et 84 ans, et 4 patients (2 %) avaient 85 ans et plus. La proportion de patients ayant présenté des événements indésirables (EI) était similaire chez les patients âgés de 65 ans et ceux âgés de 65 ans. Les patients âgés de 65 ans étaient plus susceptibles de développer des évènements indésirables graves (EIGs) et des EIs à l’origine de l’arrêt de cobimetinib que les patients âgés de 65 ans.
Population pédiatrique
La sécurité de Cotellic n’a pas été complètement établie chez les enfants et les adolescents. La sécurité de Cotellic a été évaluée dans le cadre d'une étude multicentrique, ouverte, d'escalade de dose, menée chez 55 patients pédiatriques âgés de 2 à 17 ans atteints de tumeurs solides. Le profil de sécurité de Cotellic chez ces patients était comparable à celui de la population adulte (voir rubrique 5.2).
Insuffisants rénaux
Aucune étude pharmacocinétique n’a été menée chez des patients présentant une insuffisance rénale. Aucune adaptation posologique n’est recommandée chez les patients présentant une insuffisance rénale légère à modérée, sur la base de l’analyse pharmacocinétique de population. Les données chez les patients présentant une insuffisance rénale sévère sont limitées. Cotellic doit être utilisé avec prudence chez les patients présentant une insuffisance rénale sévère.
Insuffisants hépatiques
Aucune adaptation posologique n’est recommandée chez des patients présentant une insuffisance hépatique (voir rubrique 5.2).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/15/1048/001
10. DATE DE MISE À JOUR DU TEXTE
18 mars 2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3383593 | COTELLIC 20MG COMP PELL 63 X 20MG | L01EE02 | - | € 5474 | Oui | - | - |