![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Kerendia 10 mg comprimés pelliculés
Kerendia 20 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Kerendia 10 mg comprimés pelliculés
Chaque comprimé pelliculé contient 10 mg de finérénone.
Excipient à effet notoire :
Chaque comprimé pelliculé contient 45 mg de lactose (sous forme monohydratée), voir rubrique 4.4.
Kerendia 20 mg comprimés pelliculés
Chaque comprimé pelliculé contient 20 mg de finérénone.
Excipient à effet notoire :
Chaque comprimé pelliculé contient 40 mg de lactose (sous forme monohydratée), voir rubrique 4.4.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé)
Kerendia 10 mg comprimés pelliculés
Comprimé pelliculé de couleur rose, de forme oblongue ovale, mesurant 10 mm de longueur et 5 mm de largeur, portant l’inscription « 10 » sur une face et « FI » sur l’autre.
Kerendia 20 mg comprimés pelliculés
Comprimé pelliculé de couleur jaune, de forme oblongue ovale, mesurant 10 mm de longueur et 5 mm de largeur, portant l’inscription « 20 » sur une face et « FI » sur l’autre.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Kerendia est indiqué, chez l’adulte, pour le traitement de la maladie rénale chronique (avec albuminurie) associée à un diabète de type 2.
Pour les résultats d’études concernant les événements rénaux et cardiovasculaires, voir rubrique 5.1.
4.2 Posologie et mode d’administration
Posologie
La dose cible recommandée est de 20 mg de finérénone une fois par jour.
La dose maximale recommandée est de 20 mg de finérénone une fois par jour.
Instauration du traitement
Le taux de potassium sérique et le débit de filtration glomérulaire estimé (DFGe) doivent être mesurés pour déterminer si le traitement par finérénone peut être instauré et pour déterminer la dose à l’initiation.
Si le taux de potassium sérique est ≤ 4,8 mmol/L, le traitement par finérénone peut être instauré. Pour la surveillance du potassium sérique, voir le paragraphe « Poursuite du traitement » ci-dessous.
Si le taux de potassium sérique est compris entre > 4,8 et 5,0 mmol/L, l’instauration du traitement par finérénone peut être envisagée, avec une surveillance supplémentaire du potassium sérique au cours des 4 premières semaines, selon les caractéristiques et les taux de potassium sérique du patient (voir rubrique 4.4).
Si le taux de potassium sérique est > 5,0 mmol/L, le traitement par finérénone ne doit pas être instauré (voir rubrique 4.4).
La dose recommandée de finérénone, à l’initiation, dépend du DFGe, comme indiqué dans le tableau 1.
Tableau 1 : Instauration du traitement par finérénone et dose recommandée à l’initiation
DFGe (mL/min/1,73 m2) | Dose initiale (une fois par jour) |
≥ 60 | 20 mg |
≥ 25 à < 60 | 10 mg |
< 25 | Instauration non recommandée |
Poursuite du traitement
Le taux de potassium sérique et le DFGe doivent être de nouveau mesurés 4 semaines après l’instauration ou la reprise du traitement par finérénone ou une augmentation de la dose (voir le tableau 2 pour déterminer si le traitement par finérénone peut être poursuivi et si un ajustement de la dose est nécessaire).
Par la suite, le potassium sérique doit être de nouveau mesuré à intervalles réguliers et au besoin, en fonction des caractéristiques et des taux de potassium sérique du patient.
Voir rubriques 4.4 et 4.5 pour plus d’informations.
Tableau 2 : Poursuite du traitement par finérénone et ajustement de la dose
| Dose actuelle de finérénone (une fois par jour) | ||
10 mg | 20 mg | ||
Taux de potassium sérique actuel (mmol/L) | ≤ 4,8 | Augmenter la dose de finérénone à 20 mg une fois par jour* | Poursuivre à la dose de 20 mg une fois par jour. |
> 4,8 à 5,5 | Poursuivre à la dose de 10 mg une fois par jour. | Poursuivre à la dose de 20 mg une fois par jour. | |
> 5,5 | Interrompre le traitement par finérénone. | Interrompre le traitement par finérénone. | |
* Maintenir la dose de 10 mg une fois par jour si le DFGe a diminué de plus de 30 % par rapport à la dernière mesure.
Dose oubliée
Le patient doit prendre la dose oubliée dès qu’il se rend compte de son oubli, mais uniquement au cours de la même journée.
Le patient ne doit pas prendre 2 doses pour compenser la dose oubliée.
Populations particulières
Patients âgés
Aucun ajustement de dose n’est nécessaire chez les patients âgés (voir rubrique 5.2).
Insuffisance rénale
Instauration du traitement
Chez les patients dont le DFGe est < 25 mL/min/1,73 m2, le traitement par finérénone ne doit pas être instauré compte tenu des données cliniques limitées (voir rubriques 4.4 et 5.2).
Poursuite du traitement
Chez les patients dont le DFGe est ≥ 15 mL/min/1,73 m2, le traitement par finérénone peut être poursuivi avec des ajustements de dose en fonction du taux de potassium sérique. Le DFGe doit être mesuré 4 semaines après l’instauration du traitement pour déterminer si la dose initiale peut être augmentée afin d’atteindre la dose quotidienne recommandée de 20 mg (voir le paragraphe « Poursuite du traitement » dans la rubrique « Posologie » et le tableau 2).
Compte tenu des données cliniques limitées, le traitement par finérénone doit être arrêté chez les patients ayant évolué vers une insuffisance rénale terminale (DFGe < 15 mL/min/1,73 m2) (voir rubrique 4.4).
Insuffisance hépatique
Patients présentant
- une insuffisance hépatique sévère :
Le traitement par finérénone ne doit pas être instauré (voir rubriques 4.4 et 5.2). Aucune donnée n’est disponible.
- une insuffisance hépatique modérée :
Aucun ajustement de la dose initiale n’est nécessaire. Une surveillance plus étroite du potassium sérique, qui sera adaptée en fonction des caractéristiques du patient, doit être envisagée (voir rubriques 4.4 et 5.2).
- une insuffisance hépatique légère :
Aucun ajustement de la dose initiale n’est nécessaire.
Médicaments concomitants
Chez les patients prenant de la finérénone de manière concomitante avec des inhibiteurs faibles ou modérés du CYP3A4, des suppléments de potassium, du triméthoprime ou l’association triméthoprime/sulfaméthoxazole, une surveillance plus étroite du potassium sérique, qui sera adaptée en fonction des caractéristiques du patient, doit être envisagée (voir rubrique 4.4). Les décisions relatives au traitement par finérénone doivent être prises conformément au tableau 2 (« Poursuite du traitement » dans la rubrique « Posologie »).
Une interruption temporaire du traitement par finérénone peut être nécessaire si le patient doit prendre du triméthoprime ou l’association triméthoprime/sulfaméthoxazole. Voir rubriques 4.4 et 4.5 pour plus d’informations.
Poids corporel
Aucun ajustement de dose en fonction du poids corporel n’est nécessaire (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de la finérénone chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale
Les comprimés peuvent être pris avec un verre d’eau, avec ou sans aliment (voir rubrique 5.2).
Les comprimés ne doivent pas être pris avec du pamplemousse ou du jus de pamplemousse (voir rubrique 4.5).
Écrasement des comprimés
Pour les patients incapables d’avaler les comprimés entiers, les comprimés de Kerendia peuvent être écrasés et mélangés à de l’eau ou à des aliments mous, tels que de la compote de pommes, immédiatement avant la prise par voie orale (voir rubrique 5.2).
4.3 Contre-indications
- Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
- Traitement concomitant par des inhibiteurs puissants du CYP3A4 (voir rubrique 4.5), p. ex.,
- l’itraconazole
- le kétoconazole
- le ritonavir
- le nelfinavir
- le cobicistat
- la clarithromycine
- la télithromycine
- la néfazodone
- Maladie d’Addison
4.8 Effets indésirables
Résumé du profil de sécurité
L’effet indésirable le plus fréquemment rapporté pendant le traitement par finérénone était l’hyperkaliémie (14,0 %). Voir le paragraphe ci-dessous « Description de certains effets indésirables, Hyperkaliémie » et la rubrique 4.4.
Tableau des effets indésirables
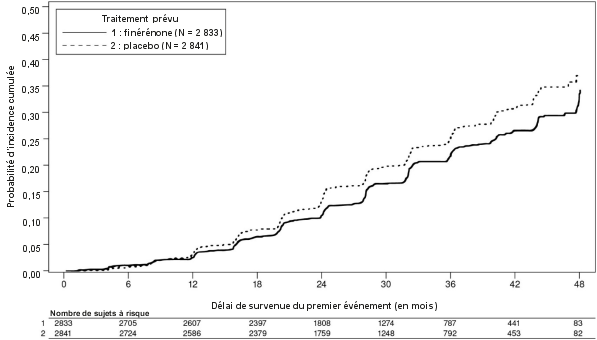
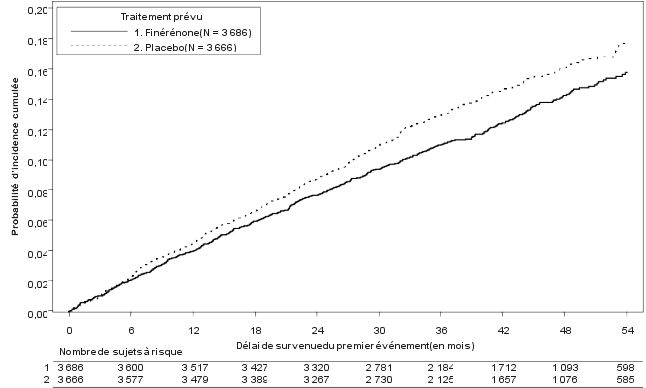
La sécurité de la finérénone chez les patients atteints de maladie rénale chronique (MRC) et de diabète de type 2 (DT2) a été évaluée dans 2 études pivots de phase III, FIDELIO‑DKD (néphropathie diabétique) et FIGARO‑DKD. Dans l’étude FIDELIO‑DKD, 2 827 patients ont reçu la finérénone (10 ou 20 mg une fois par jour) pendant une durée de traitement moyenne de 2,2 ans. Dans l’étude FIGARO‑DKD, 3 683 patients ont reçu la finérénone (10 ou 20 mg une fois par jour) pendant une durée de traitement moyenne de 2,9 ans.
Les effets indésirables observés sont répertoriés dans le tableau 3. Ils sont classés selon les classes de systèmes ou d’organes de la base de données MedDRA et par fréquence.
Les effets indésirables sont regroupés en fonction de leur fréquence, par ordre de gravité décroissante.
Les fréquences sont définies comme suit :
Très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3 : Effets indésirables
Classe de systèmes ou d’organes | Très fréquent | Fréquent | |
Troubles du métabolisme et de la nutrition | Hyperkaliémie | Hyponatrémie |
|
Affections vasculaires |
| Hypotension |
|
Affections de la peau et du tissus sous-cutané |
| Prurit |
|
Investigations |
| Débit de filtration glomérulaire diminué | Hémoglobine diminuée |
Description de certains effets indésirables
Hyperkaliémie
Dans les données poolées des études FIDELIO-DKD et FIGARO-DKD des événements d’hyperkaliémie ont été rapportés chez 14,0 % des patients traités par finérénone contre 6,9 % des patients ayant reçu le placebo. Une augmentation de 0,17 mmol/L du taux de potassium sérique moyen, par rapport à l’inclusion, a été observée au cours du premier mois de traitement dans le groupe finérénone comparé au groupe placebo, qui s’est stabilisée par la suite. Chez les patients traités par finérénone, la majorité des événements d’hyperkaliémie étaient d’intensité légère à modérée et se sont ensuite résolus. Les événements graves d’hyperkaliémie ont été rapportés plus fréquemment dans le groupe finérénone (1,1 %) que dans le groupe placebo (0,2 %). Des concentrations de potassium sérique > 5,5 mmol/L et > 6,0 mmol/L ont été rapportées chez 16,8 % et 3,3 % des patients traités par finérénone et chez 7,4 % et 1,2 % des patients ayant reçu le placebo, respectivement.
L’hyperkaliémie a entraîné l’arrêt définitif du traitement chez 1,7 % des patients traités par finérénone contre 0,6 % des patients du groupe placebo. Les hospitalisations pour hyperkaliémie dans le groupe finérénone étaient de 0,9 % contre 0,2 % dans le groupe placebo.
Pour des recommandations précises, voir les rubriques 4.2 et 4.4.
Hypotension
Dans les données poolées des études FIDELIO-DKD et FIGARO-DKD, des événements d’hypotension ont été rapportés chez 4,6 % des patients traités par finérénone contre 3,0 % des patients ayant reçu le placebo. Chez 3 patients (< 0,1 %), le traitement par finérénone a été arrêté définitivement en raison de l’hypotension. Les hospitalisations pour hypotension ont été identiques chez les patients recevant la finérénone ou le placebo (< 0,1 %).
Chez les patients traités par finérénone, la majorité des événements d’hypotension étaient d’intensité légère ou modérée et se sont ensuite résolus. La pression artérielle systolique moyenne a diminué de 2‑4 mm Hg et la pression artérielle diastolique moyenne a diminué de 1‑2 mm Hg au 1er mois, restant stables par la suite.
Hyperuricémie
Dans les données poolées des études FIDELIO-DKD et FIGARO-DKD, des événements d’hyperuricémie ont été rapportés chez 5,1 % des patients traités par finérénone contre 3,9 % des patients ayant reçu le placebo. Tous les événements observés étaient non graves et ils n’ont pas entraîné d’arrêt définitif du traitement chez les patients ayant reçu la finérénone. Une augmentation de 0,3 mg/dL du taux sérique moyen d’acide urique, par rapport à l’inclusion, a été observée dans le groupe finérénone comparé au groupe placebo jusqu’au 16e mois, qui s’est ensuite atténuée au fil du temps. S’agissant des événements de goutte rapportés, aucune différence n’a été observée entre le groupe finérénone et le groupe placebo (3,0 %).
Débit de filtration glomérulaire (DFG) diminué
Dans les données poolées des études FIDELIO-DKD et FIGARO-DKD, des événements de diminution du DFG ont été rapportés chez 5,3 % des patients traités par finérénone contre 4,2 % des patients ayant reçu le placebo. Les événements de diminution du DFG ayant entraîné l’arrêt définitif du traitement ont été identiques chez les patients ayant reçu la finérénone ou le placebo (0,2 %). Les hospitalisations pour diminution du DFG ont été identiques chez les patients recevant la finérénone ou le placebo (< 0,1 %). Chez les patients traités par finérénone, la majorité des événements de diminution du DFG étaient d’intensité légère ou modérée et se sont ensuite résolus. Les patients traités par finérénone ont présenté une baisse initiale du DFGe (2 mL/min/1,73 m2 en moyenne) comparé au groupe placebo, qui s’est atténuée au fil du temps. Cette diminution semblait réversible pendant la poursuite du traitement.
Hémoglobine diminuée
Dans les données poolées des études FIDELIO-DKD et FIGARO-DKD, la finérénone était associée à une diminution absolue, corrigée par rapport au placebo, de 0,15 g/dL du taux moyen d’hémoglobine et de 0,5 % du taux moyen de l’hématocrite après 4 mois de traitement. Les anémies rapportées ont été comparables chez les patients traités par finérénone (6,5%) et ceux ayant reçu le placebo (6,1%). La fréquence des événements graves d’anémie était faible chez les patients ayant reçu la finérénone et chez ceux ayant reçu le placebo (0,5 %). Les modifications au niveau des taux de l’hémoglobine et de l’hématrocrite étaient transitoires et ont atteint des niveaux comparables à ceux observés dans le groupe ayant reçu le placebo après environ 24‑32 mois.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de la santé déclarent tout effet indésirable suspecté via
Belgique
Agence fédérale des médicaments et des produits de santé
Division Vigilance
Boîte Postale 97
1000 Bruxelles Madou
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Bayer AG
51368 Leverkusen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
Kerendia 10 mg comprimés pelliculés
EU/1/21/1616/001-005
Kerendia 20 mg comprimés pelliculés
EU/1/21/1616/006-010
10. DATE DE MISE À JOUR DU TEXTE
02/2023
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4512018 | Kerendia 10MG COMP PELL 28 | € 72,14 | - | Oui | € 12,8 | € 8,5 | |
| 4512109 | Kerendia 10MG COMP PELL 98 | € 225,81 | - | Oui | € 15,9 | € 10,5 | |
| 4512117 | Kerendia 20MG COMP PELL 98 | € 225,81 | - | Oui | € 15,9 | € 10,5 | |
| 4512125 | Kerendia 20MG COMP PELL 28 | € 72,14 | - | Oui | € 12,8 | € 8,5 |