1. DÉNOMINATION DU MÉDICAMENT
Mayzent 0,25 mg comprimés pelliculés
Mayzent 1 mg comprimés pelliculés
Mayzent 2 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Mayzent 0,25 mg comprimés pelliculés
Chaque comprimé pelliculé contient de l’acide fumarique de siponimod équivalent à 0,25 mg de siponimod.
Excipients à effet notoire
Chaque comprimé contient 59,1 mg de lactose (sous forme monohydratée) et 0,092 mg de lécithine de soja.
Mayzent 1 mg comprimés pelliculés
Chaque comprimé pelliculé contient de l’acide fumarique de siponimod équivalent à 1 mg de siponimod.
Excipients à effet notoire
Chaque comprimé contient 58,3 mg de lactose (sous forme monohydratée) et 0,092 mg de lécithine de soja.
Mayzent 2 mg comprimés pelliculés
Chaque comprimé pelliculé contient de l’acide fumarique de siponimod équivalent à 2 mg de siponimod.
Excipients à effet notoire
Chaque comprimé contient 57,3 mg de lactose (sous forme monohydratée) et 0,092 mg de lécithine de soja.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé
Mayzent 0,25 mg comprimés pelliculés
Comprimé pelliculé de couleur rouge clair, rond, biconvexe et à bords biseautés d’environ 6,1 mm de diamètre, avec le logo du laboratoire sur une face et « T » sur l’autre face.
Mayzent 1 mg comprimés pelliculés
Comprimé pelliculé de couleur blanc-violet, rond, biconvexe et à bord biseautés d’environ 6,1 mm de diamètre, avec le logo du laboratoire sur une face et « L » sur l’autre face.
Mayzent 2 mg comprimés pelliculés
Comprimé pelliculé de couleur jaune clair, rond, biconvexe et à bords biseautés d’environ 6,1 mm de diamètre, avec le logo du laboratoire sur une face et « II » sur l’autre face.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
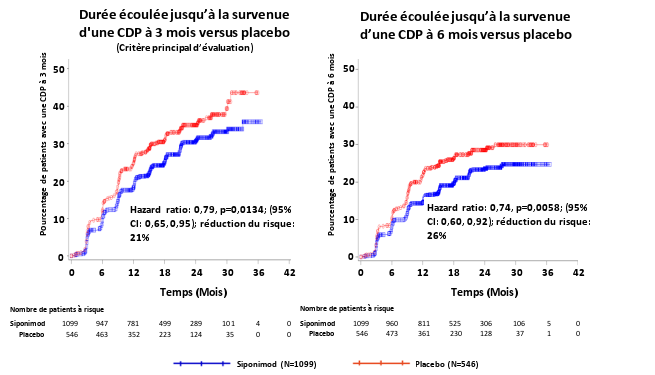
Mayzent est indiqué dans le traitement des patients adultes atteints de sclérose en plaques de forme secondairement progressive (SEP-SP) active telle que définie par des poussées ou des données d’imagerie caractéristiques d’une activité inflammatoire (voir rubrique 5.1).
4.2 Posologie et mode d'administration
Le traitement par siponimod doit être instauré et supervisé par un médecin expérimenté dans la prise en charge de la sclérose en plaques.
Avant le début du traitement, les patients doivent faire un génotypage du CYP2C9 afin de déterminer leur statut métabolique CYP2C9 (voir rubriques 4.4, 4.5 et 5.2).
Chez les patients présentant le génotype CYP2C9*3*3, le siponimod ne doit pas être utilisé (voir rubriques 4.3, 4.4 et 5.2).
Posologie
Initiation du traitement
Le traitement doit être initié avec un conditionnement de titration contenant des comprimés pour 5 jours. Le traitement débute avec l’administration d’une dose quotidienne de 0,25 mg les 1ers et 2èmes jours, puis 0,5 mg le 3ème jour, 0,75 mg le 4ème jour, et 1,25 mg le 5ème jour, afin d’atteindre la dose d’entretien de siponimod prescrite au patient à compter du 6ème jour (voir Tableau 1).
Au cours des 6 premiers jours d’initiation du traitement, la dose quotidienne recommandée doit être prise une fois par jour le matin, avec ou sans nourriture.
Tableau 1 Schéma de titration posologique pour atteindre la dose d’entretien
Titration | Dose | Schéma | Dose |
Jour 1 | 0,25 mg | 1 x 0,25 mg | TITRATION |
Jour 2 | 0,25 mg | 1 x 0,25 mg | |
Jour 3 | 0,5 mg | 2 x 0,25 mg | |
Jour 4 | 0,75 mg | 3 x 0,25 mg | |
Jour 5 | 1,25 mg | 5 x 0,25 mg | |
Jour 6 | 2 mg1 | 1 x 2 mg1 | ENTRETIEN |
1 Chez les patients présentant le génotype CYP2C9*2*3 ou *1*3, la dose d’entretien recommandée est de 1 mg une fois par jour (1 x 1 mg ou 4 x 0,25 mg) (voir ci-dessus et les rubriques 4.4 et 5.2). Une exposition additionnelle de 0,25 mg le 5ème jour ne remet pas en cause la sécurité du patient. | |||
Traitement d’entretien
Chez les patients présentant le génotype CYP2C9*2*3 ou *1*3, la dose d’entretien recommandée est de 1 mg (voir rubriques 4.4 et 5.2).
Chez les patients présentant tous les autres génotypes du CYP2C9, la dose d’entretien recommandée de siponimod est de 2 mg.
Mayzent se prend une fois par jour.
Oubli de dose(s) lors de l’initiation du traitement
En cas d’oubli de dose au cours des 6 premiers jours d’initiation du traitement, le traitement doit être réinitié avec un nouveau conditionnement de titration.
Oubli de dose après le 6ème jour
En cas d’oubli de dose, la dose prescrite doit être prise lors de la prochaine administration prévue ; la dose suivante ne doit pas être doublée.
Réinitiation du traitement d’entretien après interruption
Si le traitement d’entretien est interrompu à raison de 4 doses quotidiennes consécutives ou plus, le siponimod doit être réinitié avec un nouveau conditionnement de titration.
Populations particulières
Patients âgés
Le siponimod n’a pas été étudié chez les patients agés de 65 ans et plus. Des patients jusqu’à l’âge de 61 ans ont été inclus dans les études cliniques. Le siponimod doit être administré avec prudence chez les patients âgés en raison de l’insuffisance de données de sécurité et d’efficacité (voir rubrique 5.2).
Insuffisance rénale
D’après les études de pharmacologie clinique, aucun ajustement posologique n’est nécessaire chez les patients atteints d’insuffisance rénale (voir rubrique 5.2).
Insuffisance hépatique
Le siponimod ne doit pas être administré chez les patients atteints d’insuffisance hépatique sévère (classe C de Child-Pugh) (voir rubrique 4.3). Bien qu’aucun ajustement posologique ne soit nécessaire chez les patients atteints d’insuffisance hépatique légère ou modérée, il convient d’initier le traitement avec prudence chez ces patients (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité du siponimod chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie orale. Le siponimod se prend avec ou sans nourriture.
Les comprimés pelliculés doivent être avalés entiers avec de l’eau.
4.3 Contre-indications
- Hypersensibilité à la substance active ou à la cacahuète, au soja ou à l'un des excipients mentionnés à la rubrique 6.1.
- Syndrome d’immunodéficience.
- Antécédents de leucoencéphalopathie multifocale progressive ou de méningite à cryptocoques.
- Cancers en évolution.
- Insuffisance hépatique sévère (classe C de Child-Pugh).
- Patients ayant présenté dans les 6 derniers mois un infarctus du myocarde (IM), un angor instable, un accident vasculaire cérébral (AVC)/accident ischémique transitoire (AIT), une insuffisance cardiaque décompensée (nécessitant une hospitalisation) ou une insuffisance cardiaque de classe III/IV selon la classification New York Heart Association (NYHA) (voir rubrique 4.4).
- Patients avec des antécédents de bloc auriculo-ventriculaire (BAV) du second degré de type Mobitz II, de BAV du troisième degré, de bloc sino-auriculaire ou de maladie du sinus, en l’absence de port de pacemaker (voir rubrique 4.4).
- Patients homozygotes pour le génotype CYP2C9*3 (CYP2C9*3*3) (métaboliseurs lents).
- Pendant la grossesse et chez les femmes en âge de procréer n’utilisant pas une contraception efficace (voir rubriques 4.4 et 4.6).
4.8 Effets indésirables
Résumé du profil de sécurité
Le profil de sécurité du siponimod est basé sur les données de l’étude clinique principale. Les effets indésirables les plus fréquents identifiés dans la phase principale de l’étude A2304 étaient les céphalées (15 %) et l’hypertension (12,6 %). Les informations relatives à la sécurité issues de la phase d’extension de l’étude à long terme A2304 étaient cohérentes avec celles observées dans la phase principale.
Tableau récapitulatif des effets indésirables
Au sein de chaque classe de systèmes d’organes, les effets indésirables sont classés par fréquence, les effets les plus fréquents apparaissant en premier. Les fréquences ont été définies selon la convention suivante : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/1 000, <1/100) ; rare (≥1/10 000, <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Tableau 2 Tableau récapitulatif des effets indésirables
Infections et infestations | |
Fréquent | Zona |
Rare | Leucoencéphalopathie multifocale progressive |
Fréquence indéterminée | Méningite à cryptocoques |
Tumeurs bénignes, malignes et non précisées (dont kystes et polypes) | |
Fréquent | Nævus mélanocytaire |
Peu fréquent | Carcinome spino-cellulaire |
Affections hématologiques et du système lymphatique | |
Fréquent | Lymphopénie |
Affections du système immunitaire | |
Rare | Syndrome inflammatoire de reconstitution immune (IRIS) |
Affections du système nerveux | |
Très fréquent | Céphalées |
Fréquent | Sensation vertigineuse |
Affections oculaires | |
Fréquent | Œdème maculaire |
Affections cardiaques | |
Fréquent | Bradycardie |
Affections vasculaires | |
Très fréquent | Hypertension |
Affections gastro-intestinales | |
Fréquent | Nausées |
Affections musculo-squelettiques et systémiques | |
Fréquent | Douleurs des extrémités |
Troubles généraux et anomalies au site d'administration | |
Fréquent | Œdème périphérique |
Investigations | |
Très fréquent | Élévation des paramètres biologiques hépatiques |
Fréquent | Diminution des paramètres fonctionnels respiratoires |
Description d’effets indésirables sélectionnés
Infections
Dans l’étude clinique de phase III conduite chez des patients atteints de SEP-SP, le taux global d’infections était comparable entre les patients sous siponimod et ceux sous placebo (49,0 % versus 49,1 %, respectivement). Cependant, une augmentation de la fréquence des zonas a été rapportée sous siponimod (2,5 %) par rapport au placebo (0,7 %).
Des cas de méningite ou méningo-encéphalite causés par des virus varicelle-zona sont survenus à tout moment lors du traitement par siponimod. Des cas de méningite à cryptocoques (MC) ont également été rapportés avec le siponimod (voir rubrique 4.4).
Œdème maculaire
Un œdème maculaire a été rapporté plus fréquemment chez les patients recevant le siponimod (1,8 %) que chez ceux recevant le placebo (0,2 %). Même si la majorité des cas est survenue au cours des 3 à 4 premiers mois de traitement par siponimod, des cas ont également été rapportés après 6 mois de traitement par siponimod (voir rubrique 4.4). Certains patients ont présenté une vision trouble ou une diminution de l’acuité visuelle, mais d’autres étaient asymptomatiques et ont été diagnostiqués lors d’un examen ophtalmologique de routine. L’œdème maculaire a généralement regressé ou s’est résorbé spontanément après l’arrêt du traitement. Le risque de récidive après réintroduction du traitement n’a pas été évalué.
Bradyarythmie
L’initiation du traitement par siponimod entraîne une diminution transitoire de la fréquence cardiaque et peut également être associé à des retards de conduction auriculo-ventriculaire (voir rubrique 4.4). Une bradycardie a été rapportée chez 6,2 % des patients traités par siponimod versus 3,1 % des patients sous placebo et un bloc auriculo-ventriculaire (BAV) a été rapporté chez 1,7 % des patients traités par siponimod versus 0,7 % des patients sous placebo (voir rubrique 4.4).
La diminution maximale de la fréquence cardiaque est observée dans les 6 heures suivant l’administration.
Une diminution dose-dépendante transitoire de la fréquence cardiaque a été observée lors de la phase initiale de titration et s’est stabilisée aux doses ≥5 mg. Des événements bradyarythmiques (BAV et pauses sinusales) ont été détectés avec une incidence plus élevée sous traitement par siponimod que sous placebo.
La plupart des BAV et des pauses sinusales sont survenus au-dessus de la dose thérapeutique de 2 mg, avec une incidence notablement plus élevée en l’absence de titration qu’avec titration de la dose.
La diminution de la fréquence cardiaque induite par le siponimod peut être inversée par l’administration d’atropine ou d’isoprénaline.
Fonction hépatique
Des élévations des enzymes hépatiques (principalement des élévations des taux d’ALAT) ont été rapportées chez les patients atteints de SEP et traités par siponimod. Dans l’étude de phase III conduite chez des patients atteints de SEP-SP, les élévations des paramètres hépatiques ont été plus fréquemment observées chez les patients sous siponimod (11,3 %) que chez ceux sous placebo (3,1 %), principalement en raison d’élévations des transaminases hépatiques (ALAT/ASAT) et des gammaGT. La majorité de ces élévations se sont produites dans les 6 premiers mois du traitement. Les taux d’ALAT sont revenus à la normale environ 1 mois après l’arrêt du traitement par siponimod (voir rubrique 4.4).
Pression artérielle
Dans l’étude clinique de phase III conduite chez des patients atteints de SEP-SP, une hypertension a été rapportée plus fréquemment chez les patients sous siponimod (12,6 %) que chez ceux sous placebo (9,0 %). Le traitement par siponimod a entraîné une augmentation de la pression artérielle systolique et diastolique rapidement après le début du traitement, l’effet étant maximal après environ 6 mois de traitement (systolique 3 mmHg, diastolique 1,2 mmHg) et se stabilisant par la suite. Cet effet a persisté avec la poursuite du traitement.
Crises épileptiques
Dans l’étude clinique de phase III conduite chez des patients atteints de SEP-SP, des crises épileptiques ont été rapportés chez 1,7 % des patients traités par siponimod contre 0,4 % sous placebo.
Effets respiratoires
Des diminutions mineures des valeurs du volume expiratoire maximal par seconde (VEMS) et de la capacité de diffusion du monoxyde de carbone (DLCO) ont été observées avec le traitement par siponimod. Dans l’étude clinique de phase III conduite chez des patients atteints de SEP-SP, les variations moyennes du VEMS par rapport aux valeurs initiales étaient de -0,1 L dans le groupe siponimod après 3 mois et 6 mois de traitement, alors qu’aucune variation n’était observée dans le groupe placebo. Ces observations étaient légèrement plus élevées (variations moyennes du VEMS d’environ 0,15 L par rapport aux valeurs initiales) chez les patients présentant des troubles respiratoires tels qu’une bronchopneumopathie chronique obstructive (BPCO) ou un asthme et traités par siponimod. Sous traitement chronique, cette réduction ne s’est pas traduite par des événements indésirables cliniquement significatifs et n’a pas été associée à une augmentation des signalements de toux ou de dyspnée (voir rubrique 5.1).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via:
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet: www.guichet.lu/pharmacovigilance

7. TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHÉ
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irlande
8. NUMÉRO(S) D'AUTORISATION DE MISE SUR LE MARCHÉ
Mayzent 0,25 mg comprimés pelliculés
EU/1/19/1414/001
EU/1/19/1414/002
EU/1/19/1414/004
Mayzent 1 mg comprimés pelliculés
EU/1/19/1414/007
EU/1/19/1414/008
Mayzent 2 mg comprimés pelliculés
EU/1/19/1414/003
EU/1/19/1414/005
EU/1/19/1414/006
10. DATE DE MISE À JOUR DU TEXTE
06.05.2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l'Agence européenne des médicaments https://www.ema.europa.eu.
1
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4166732 | MAYZENT 0,25MG COMP PELL 12 | L04AA42 | € 278,92 | € 244,45 | Oui | € 12,8 | € 8,5 |
| 4166724 | MAYZENT 0,25MG COMP PELL 120 | L04AA42 | € 1616,1 | € 1466,79 | Oui | - | - |
| 4166716 | MAYZENT 2,00MG COMP PELL 28 | L04AA42 | € 1511,6 | € 1369 | Oui | € 12,8 | € 8,5 |
| 4506754 | MAYZENT 1,00MG COMP PELL 28 | L04AA42 | € 1511,6 | - | Oui | € 12,8 | € 8,5 |