RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Lokelma 5 g, poudre pour suspension buvable
Lokelma 10 g, poudre pour suspension buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Lokelma 5 g, poudre pour suspension buvable
Chaque sachet contient 5 g de cyclosilicate de zirconium sodique
Chaque sachet de 5 g contient approximativement 400 mg de sodium
Lokelma 10 g, poudre pour suspension buvable
Chaque sachet contient 10 g de cyclosilicate de zirconium sodique
Chaque sachet de 10 g contient approximativement 800 mg de sodium
3. FORME PHARMACEUTIQUE
Poudre pour suspension buvable.
Poudre blanche à grise.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Lokelma est indiqué dans le traitement de l’hyperkaliémie chez l’adulte (voir rubriques 4.4 et 5.1).
4.2 Posologie et mode d’administration
Posologie
Phase de correction
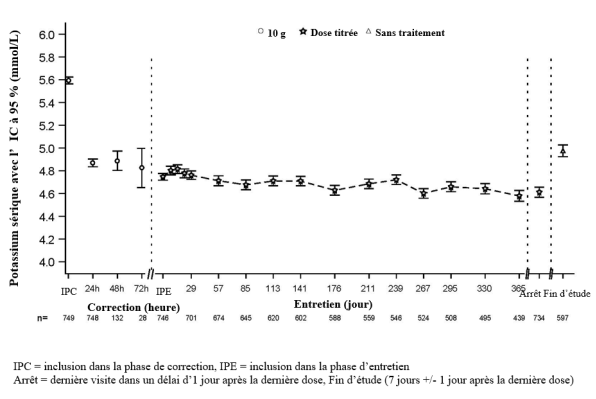
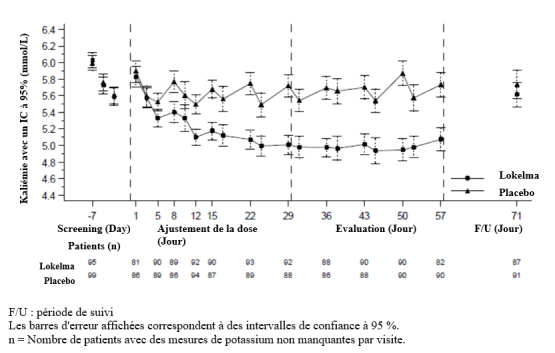
La dose initiale recommandée de Lokelma est de 10 g, administrée trois fois par jour par voie orale sous forme d’une suspension dans l’eau. Lorsque la normokaliémie est atteinte, la posologie d’entretien doit être suivie (voir ci-dessous).
En règle générale, la normokaliémie est atteinte en 24 à 48 heures. Si les patients présentent toujours une hyperkaliémie après 48 heures de traitement, la même posologie peut être poursuivie pendant 24 heures supplémentaires.
Si la normokaliémie n’est pas atteinte après 72 heures de traitement, d’autres approches thérapeutiques doivent être envisagées.
Phase d’entretien
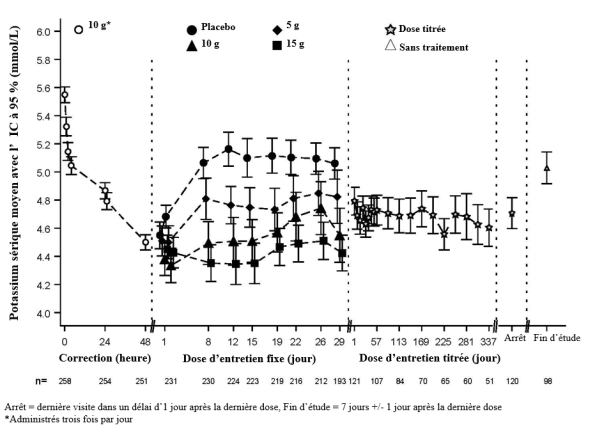
Lorsque la normokaliémie est obtenue, la dose minimale efficace de Lokelma pour prévenir la récidive de l’hyperkaliémie doit être établie. Une dose initiale de 5 g une fois par jour est recommandée, avec une augmentation possible jusqu’à 10 g une fois par jour, ou une diminution jusqu’à 5 g un jour sur deux, selon les besoins, afin de maintenir un taux normal de potassium. La dose quotidienne à utiliser pour le traitement d’entretien ne doit pas dépasser 10 g une fois par jour.
La kaliémie doit être surveillée régulièrement pendant le traitement (voir rubrique 4.4).
Oubli d’une dose
Si un patient oublie une dose, il doit être informé de prendre la dose suivante à l’heure habituellement prévue.
Populations particulières
Patients présentant une insuffisance rénale
Aucune modification des doses normales n’est nécessaire chez les patients présentant une insuffisance rénale qui ne sont pas sous hémodialyse chronique.
Pour les patients sous dialyse, Lokelma ne doit être administré que les jours sans dialyse. La dose initiale recommandée est de 5 g une fois par jour. Pour obtenir une normokaliémie (4,0-5,0 mmol/L), la dose peut être augmentée ou diminuée chaque semaine en fonction de la valeur de kaliémie en pré-dialyse après l’intervalle interdialytique long (LIDI = long inter‑dialytic interval). La dose peut être ajustée à une semaine d’intervalle par incrémentation de 5 g jusqu'à 15 g une fois par jour les jours sans dialyse. Il est recommandé de surveiller la kaliémie chaque semaine pendant l’ajustement de la dose ; une fois la normokaliémie obtenue, le potassium doit être surveillé régulièrement (par exemple, mensuellement, ou plus fréquemment sur la base d'un jugement clinique, y compris les changements dans le potassium alimentaire ou les médicaments affectant la kaliémie).
Patients présentant une insuffisance hépatique
Aucune modification des doses normales n’est nécessaire chez les patients présentant une insuffisance hépatique.
Sujets âgés
Il n’existe aucune recommandation spécifique sur la dose et l’administration pour cette population.
Population pédiatrique
La sécurité d’emploi et l’efficacité de Lokelma chez les enfants et les adolescents (âgés < 18 ans) n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale.
Le contenu entier du(des) sachet(s) doit être vidé dans un verre contenant environ 45 ml d’eau et être correctement remué. Le liquide insipide doit être bu pendant qu’il est encore trouble. La poudre ne se dissoudra pas. Si la poudre se dépose, l’eau doit de nouveau être remuée et bue. Si nécessaire, rincer le verre avec plus d’eau pour s’assurer de la prise complète du contenu.
La suspension peut être prise au cours ou en dehors des repas.
4.3 Contre-indications
Hypersensibilité à la substance active.
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés ont été de l’hypokaliémie (4,1 %) et des événements liés à des œdèmes (5,7 %).
Dans deux études cliniques avec une exposition à Lokelma en phase ouverte jusqu’à 1 an chez 874 patients, les effets indésirables suivants ont été rapportés comme reliés au produit par les investigateurs : événements gastro-intestinaux [constipation (2,9 %), nausées (1,6 %), diarrhée (0,9 %), douleur / distension abdominale (0,5 %) et vomissements (0,5 %)] ; et réactions d’hypersensibilité [éruption cutanée (0,3 %) et prurit (0,1 %)]. Ces événements étaient d’intensité légère à modérée, aucun n’a été signalé comme grave et ils se sont généralement résolus alors que le patient poursuivait le traitement. Compte tenu de la conception de l’étude en phase ouverte, un lien de causalité entre ces événements et Lokelma ne peut pas être établi.
Dans les études cliniques réalisées dans les pays de population à prédominance asiatique, une constipation avec une fréquence estimée à 8,9 % est survenue chez les patients non dialysés recevant du Lokelma et elle a été résolue avec un ajustement de la dose ou un arrêt du traitement.
Dans une analyse groupée de trois études cliniques contrôlées versus placebo portant sur Lokelma chez des patients non dialysés, certains patients présentant une insuffisance cardiaque préexistante ont connu une aggravation de leur insuffisance cardiaque, survenant à une fréquence de 13,6 % (30/220) sous Lokelma et de 5,7 % (12/209) sous placebo. La plupart des cas ont été résolus par une prise en charge clinique appropriée sans arrêt de Lokelma (voir rubrique 4.4).
Liste tabulée des effets indésirables
Le profil de sécurité de Lokelma a été évalué dans des essais cliniques incluant 1760 patients dont 507 patients exposés pendant une année.
Les effets indésirables observés dans les essais contrôlés et lors de la surveillance en post-commercialisation sont présentés dans le Tableau 1. Les effets indésirables mentionnés ci‑dessous sont classés par fréquence et par classe de systèmes d’organes (SOC).
La convention suivante a été utilisée pour décrire la fréquence des effets indésirables : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), fréquence non déterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1. Liste des effets indésirables observés dans les études cliniques et lors de la surveillance en post-commercialisation
Classe de systèmes d’organes | Très fréquent | Fréquent |
Troubles du métabolisme et de la nutrition |
| Hypokaliémie |
Troubles gastro-intestinaux |
| Constipation |
Troubles généraux et anomalies au site d’administration |
| Événements liés à des œdèmes |
Affections cardiaques | Aggravation d’une insuffisance cardiaque préexistante |
|
Description d’effets indésirables sélectionnés
Hypokaliémie
Dans les essais cliniques, 4,1 % des patients sous Lokelma ont développé une hypokaliémie avec une valeur de la kaliémie inférieure à 3,5 mmol/L, qui a été résolue par un ajustement de la dose ou un arrêt de Lokelma.
Événements liés à des œdèmes
Les événements liés à des œdèmes, incluant rétention hydrique, œdème généralisé, hypervolémie, œdème localisé, œdème, œdème périphérique et gonflement périphérique, ont été rapportés chez 5,7 % des patients sous Lokelma. Les événements ont été observés durant la phase d’entretien uniquement et étaient plus fréquemment observés chez les patients traités avec 15 g. Jusqu’à 53 % des patients ont été pris en charge en initiant un diurétique ou en ajustant la dose de diurétique ; les autres n’ont pas nécessité de traitement.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de la santé déclarent tout effet indésirable suspecté via :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance :
Site internet : www.notifieruneffetindesirable.be
e-mail : adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
AstraZeneca AB
SE-151 85 Södertälje
Suède
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1173/001
EU/1/17/1173/002
EU/1/17/1173/003
EU/1/17/1173/004
EU/1/17/1173/007
EU/1/17/1173/009
EU/1/17/1173/010
EU/1/17/1173/012
10. DATE DE MISE À JOUR DU TEXTE
07/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4235586 | LOKELMA 5G PDR POUR SUSP ORALE SACH 30 | V03AE10 | € 323,35 | - | Oui | € 2 | € 1 |
| 4235669 | LOKELMA 10G PDR POUR SUSP ORALE SACH 30 | V03AE10 | € 323,35 | - | Oui | € 2 | € 1 |