1. DENOMINATION DU MEDICAMENT
Xarelto 2,5 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient 2,5 mg de rivaroxaban.
Excipient à effet notoire
Chaque comprimé pelliculé contient 33,92 mg de lactose (sous forme monohydratée), voir rubrique 4.4.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé).
Comprimé jaune clair, rond, biconvexe (diamètre de 6 mm, rayon de courbure de 9 mm) marqué de la croix BAYER sur une face et sur l’autre face du nombre « 2,5 » et d’un triangle.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Xarelto, co-administré avec de l’acide acétylsalicylique (AAS) seul ou avec de l’AAS plus du clopidogrel ou de la ticlopidine, est indiqué pour la prévention des événements athérothrombotiques chez les patients adultes suite à un syndrome coronarien aigu (SCA) avec élévation des biomarqueurs cardiaques (voir rubriques 4.3, 4.4 et 5.1).
Xarelto, co-administré avec de l’acide acétylsalicylique (AAS), est indiqué pour la prévention des événements athérothrombotiques chez les patients adultes présentant une maladie coronarienne (MC) ou une maladie artérielle périphérique (MAP) symptomatique à haut risque d’événements ischémiques.
4.2 Posologie et mode d’administration
Posologie
La dose recommandée est de deux prises par jour de 2,5 mg.
- SCA
Les patients sous Xarelto 2,5 mg deux fois par jour doivent également prendre une dose quotidienne de 75 - 100 mg d’AAS ou une dose quotidienne de 75 - 100 mg d’AAS en complément d’une dose quotidienne de 75 mg de clopidogrel ou d’une dose quotidienne standard de ticlopidine.
L’intérêt du traitement doit être régulièrement évalué au cas par cas après évaluation du risque d’événements ischémiques par rapport au risque de saignement. L’expérience étant limitée à 24 mois, une prolongation du traitement au-delà de 12 mois doit être définie au cas par cas (voir rubrique 5.1).
Le traitement par Xarelto doit être débuté dès que possible après la phase de stabilisation du SCA (comprenant également les procédures de revascularisation) ; au plus tôt 24 heures après l’admission à l’hôpital et au moment où le patient ne requière plus de traitement anticoagulant dans le cadre du SCA.
- MC/MAP
Les patients sous Xarelto 2,5 mg deux fois par jour doivent également prendre une dose quotidienne de 75 ‑ 100 mg d’AAS.
Chez les patients ayant bénéficié d’une procédure de revascularisation réussie (chirurgicale ou endovasculaire, procédures hybrides incluses) d’un membre inférieur suite à une MAP symptomatique, le traitement ne doit pas être instauré avant que l’hémostase soit obtenue (voir rubrique 5.1).
La durée du traitement sera déterminée au cas par cas pour chaque patient de façon régulière et elle tiendra compte du risque d’événements thrombotiques par rapport au risque de saignements.
- SCA, MC/MAP
Administration concomitante avec un traitement antiplaquettaire
Chez les patients présentant un événement thrombotique aigu ou ayant subi une procédure vasculaire et nécessitant une bithérapie antiplaquettaire, la poursuite de Xarelto 2,5 mg deux fois par jour devra être évaluée en fonction du type d’événement ou de procédure et du schéma posologique antiplaquettaire.
La sécurité et l’efficacité de Xarelto 2,5 mg deux fois par jour en association avec une bithérapie antiplaquettaire ont été étudiées chez des patients :
- ayant récemment présenté un SCA, en association avec l’ASS plus clopidogrel/ticlopidine (voir rubrique 4.1), et
- ayant récemment bénéficié d’une procédure de revascularisation d’un membre inférieur suite à une MAP symptomatique, en association avec l’AAS et, s’il y a lieu, avec du clopidogrel sur une courte durée (voir rubriques 4.4 et 5.1)
Oubli d’une dose
En cas d’oubli d’une dose, le patient doit poursuivre le traitement normalement en prenant la dose recommandée suivante à l’heure habituelle. La dose ne doit pas être doublée pour compenser une dose oubliée.
Relais des anti-vitamine K (AVK) par Xarelto
Lors du passage des AVK à Xarelto, les valeurs du rapport international normalisé (INR) pourraient être faussement élevées suite à la prise de Xarelto. L’INR ne convient pas pour mesurer l’activité anticoagulante de Xarelto et ne doit donc pas être utilisé (voir rubrique 4.5).
Relais de Xarelto par les anti-vitamine K (AVK)
Il existe un risque d’anticoagulation inadéquate lors du relais de Xarelto par les AVK. Une anticoagulation continue adéquate doit être assurée lors du relais par un autre anticoagulant. Il est à noter que Xarelto peut contribuer à l’élévation de l’INR.
En cas de relais de Xarelto par un AVK, l’AVK doit être administré conjointement jusqu’à ce que l’INR soit ≥ 2,0. Lors des deux premiers jours du relais, l’AVK doit être utilisé à sa posologie initiale standard, puis la posologie doit être adaptée sur la base des mesures de l’INR. Lorsque les patients reçoivent simultanément Xarelto et l’AVK, l’INR doit être mesuré à partir de 24 heures après la dernière dose de Xarelto et avant la dose suivante. Une fois le traitement par Xarelto interrompu, des mesures fiables de l’INR ne peuvent être obtenues que 24 heures après la dernière dose de Xarelto (voir rubriques 4.5 et 5.2).
Relais des anticoagulants parentéraux par Xarelto
Chez les patients recevant un anticoagulant parentéral, arrêtez l’anticoagulant parentéral et initiez le traitement par Xarelto 0 à 2 heures avant l’heure à laquelle l’administration suivante du médicament parentéral (héparines de bas poids moléculaire, par ex.) aurait été prévue ou au moment de l’arrêt du médicament parentéral en cas d’administration continue (héparine non fractionnée intraveineuse, par ex.).
Relais de Xarelto par les anticoagulants parentéraux
Arrêtez Xarelto et administrez la première dose d’anticoagulant parentéral à l’heure à laquelle la dose suivante de Xarelto aurait dû être prise.
Populations particulières
Insuffisance rénale
Chez les patients atteints d’insuffisance rénale sévère (clairance de la créatinine de 15 à 29 ml/min), les données cliniques sont limitées mais montrent une augmentation significative des concentrations plasmatiques du rivaroxaban. Chez ces patients, Xarelto doit donc être utilisé avec prudence. L'utilisation n’est pas recommandée chez les patients dont la clairance de la créatinine est < 15 ml/min (voir rubriques 4.4 et 5.2).
Aucun ajustement posologique n’est nécessaire chez les patients atteints d’insuffisance rénale légère (clairance de la créatinine de 50 à 80 ml/min) ou d’insuffisance rénale modérée (clairance de la créatinine de 30 à 49 ml/min) (voir rubrique 5.2).
Insuffisance hépatique
L’utilisation de Xarelto est contre-indiquée chez les patients présentant une atteinte hépatique associée à une coagulopathie et à un risque de saignement cliniquement significatif, y compris chez les patients cirrhotiques avec un score de Child Pugh classe B ou C (voir rubriques 4.3 et 5.2).
Personnes âgées
Aucun ajustement posologique (voir rubriques 4.4 et 5.2)
Le risque de saignement augmente avec l’âge (voir rubrique 4.4).
Poids
Aucun ajustement posologique (voir rubriques 4.4 et 5.2)
Sexe
Aucun ajustement posologique (voir rubrique 5.2)
Population pédiatrique
La sécurité et l’efficacité des comprimés de Xarelto 2,5 mg n’ont pas été établies chez les enfants âgés de 0 à 18 ans. Aucune donnée n’est disponible. L’utilisation des comprimés de Xarelto 2.5 mg n’est donc pas recommandée chez l'enfant de moins de 18 ans.
Mode d’administration
Xarelto est pour usage par voie orale.
Les comprimés peuvent être pris au cours ou en dehors des repas (voir rubriques 4.5 et 5.2).
Ecrasement des comprimés
Pour les patients qui sont dans l’incapacité d’avaler les comprimés entiers, le comprimé de Xarelto peut être écrasé et mélangé à de l’eau ou à de la compote de pommes, juste avant administration par voie orale.
Le comprimé écrasé peut également être administré au moyen d’une sonde gastrique (voir rubriques 5.2 et 6.6).
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Saignement évolutif cliniquement significatif.
Lésion ou maladie, si considérée comme étant à risque significatif de saignement majeur. Cela peut comprendre : ulcération gastrointestinale en cours ou récente, présence de tumeurs malignes à haut risque de saignement, lésion cérébrale ou rachidienne récente, chirurgie cérébrale, rachidienne ou ophtalmique récente, hémorragie intracrânienne récente, varices oesophagiennes connues ou suspectées, malformations artérioveineuses, anévrismes vasculaires ou anomalies vasculaires majeures intrarachidiennes ou intracérébrales.
Traitement concomitant avec tout autre anticoagulant, par exemple, héparine non-fractionnée (HNF), héparines de bas poids moléculaire (énoxaparine, daltéparine, etc), dérivés de l’héparine (fondaparinux, etc), anticoagulants oraux (warfarine, dabigatran etexilate, apixaban, etc) sauf dans des circonstances spécifiques de relais de traitement anticoagulant (voir rubrique 4.2) ou en cas d’administration d’HNF aux doses nécessaires pour le maintien de la perméabilité d’un cathéter central veineux ou artériel (voir rubrique 4.5).
Traitement concomitant du SCA avec un traitement antiplaquettaire chez les patients présentant des antécédents d’accident vasculaire cérébral (AVC) ou d’accident ischémique transitoire (AIT) (voir rubrique 4.4).
Traitement concomitant de la MC/MAP par de l’AAS chez les patients ayant déjà présenté un AVC hémorragique ou lacunaire, ou tout autre type d’AVC au cours du mois précédent (voir rubrique 4.4).
Atteinte hépatique associée à une coagulopathie et à un risque de saignement cliniquement significatif, y compris les patients cirrhotiques avec un score de Child Pugh classe B ou C (voir rubrique 5.2).
Grossesse et allaitement (voir rubrique 4.6).
4.8 Effets indésirables
Résumé du profil de sécurité
La tolérance du rivaroxaban a été évaluée dans treize études pivots de phase III (voir Tableau 1).
Au total, 69 608 patients adultes inclus dans 19 études de phase III et 488 patients pédiatriques inclus dans deux études de phase II et deux études de phase III ont été exposés au rivaroxaban.
Tableau 1 : Nombre de patients étudiés, dose quotidienne totale et durée maximale du traitement dans les études de phase III menées chez des patients adultes et pédiatriques
Indication | Nombre de patients* | Dose quotidienne totale | Durée maximale du traitement |
Prévention des évènements thromboemboliques veineux (ETEV) chez les patients adultes bénéficiant d’une chirurgie programmée de la hanche ou du genou | 6 097 | 10 mg | 39 jours |
Prévention des ETEV chez les patients présentant une affection médicale aigüe | 3 997 | 10 mg | 39 jours |
Traitement de la thrombose veineuse profonde (TVP), de l’embolie pulmonaire (EP) et prévention des récidives | 6 790 | Jours 1-21 : 30 mg | 21 mois |
Traitement des ETEV et prévention des récidives sous forme d’ETEV chez les nouveau-nés nés à terme et chez les enfants âgés de moins de 18 ans après l’instauration d’un traitement anticoagulant standard | 329 | Dose ajustée selon le poids corporel pour atteindre une exposition similaire à celle observée chez les adultes traités pour une TVP avec 20 mg de rivaroxaban une fois par jour | 12 mois |
Prévention des AVC et des embolies systémiques chez les patients atteints de fibrillation atriale non valvulaire | 7 750 | 20 mg | 41 mois |
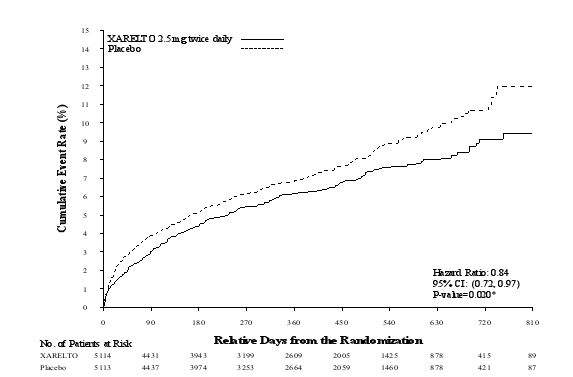
Prévention des événements athérothrombotiques suite à un syndrome coronarien aigu (SCA) | 10 225 | 5 mg ou 10 mg respectivement, co-administré avec de l’AAS ou de l’AAS associé au clopidogrel ou à la ticlopidine | 31 mois |
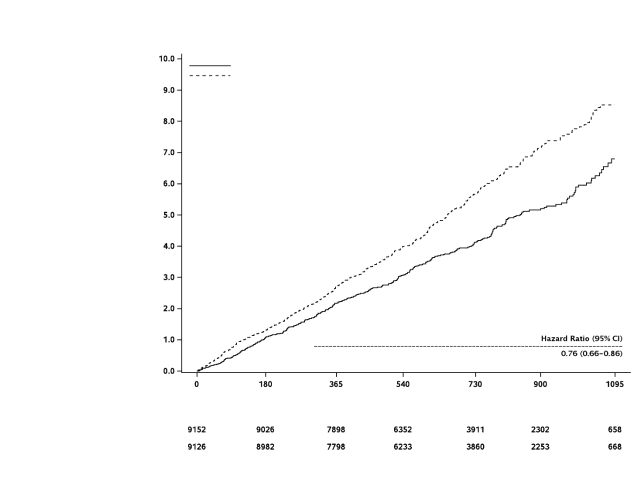
Prévention des événements athérothrombotiques chez les patients présentant une MC/MAP | 18 244 | 5 mg co-administrés avec de l’AAS ou 10 mg seuls | 47 mois |
3 256** | 5 mg co-administrés avec de l’AAS | 42 mois |
* Patients exposés à au moins une dose de rivaroxaban
** Dans l’étude VOYAGER PAD
Les effets indésirables signalés le plus fréquemment chez les patients recevant du rivaroxaban ont été les saignements (Tableau 2) (voir aussi rubrique 4.4. et « Description de certains effets indésirables » ci-dessous). Parmi les saignements signalés, les plus fréquents ont été l’épistaxis (4,5 %) et l’hémorragie du tractus gastro-intestinal (3,8 %).
Tableau 2 : Taux de survenue des saignements* et des anémies chez les patients exposés au rivaroxaban au cours des études de phase III terminées chez des patients adultes et pédiatriques
Indication | Tout saignement | Anémie |
Prévention des ETEV chez les patients adultes bénéficiant d’une chirurgie programmée de la hanche ou du genou | 6,8% des patients | 5,9% des patients |
Prévention des ETEV chez les patients présentant une affection médicale aigüe | 12,6% des patients | 2,1% des patients |
Traitement de la TVP, de l’EP et prévention des récidives | 23% des patients | 1,6% des patients |
Traitement des ETEV et prévention des récidives sous forme d’ETEV chez les nouveau-nés nés à terme et chez les enfants âgés de moins de 18 ans après l’instauration d’un traitement anticoagulant standard | 39,5 % des patients | 4,6 % des patients |
Prévention des AVC et des embolies systémiques chez les patients atteints de fibrillation atriale non valvulaire | 28 pour 100 patient-années | 2,5 pour 100 patient-années |
Prévention des événements athérothrombotiques suite à un SCA | 22 pour 100 patient-années | 1,4 pour 100 patient-années |
Prévention des événements athérothrombotiques chez les patients présentant une MC/MAP | 6,7 pour 100 patient-années | 0,15 pour 100 patient-années** |
8,38 pour 100 patient-années# | 0,74 pour 100 patient-années*** # |
* Pour toutes les études sur le rivaroxaban, tous les événements hémorragiques sont recueillis, rapportés et adjudiqués.
** Dans l’étude COMPASS, il y a une faible incidence des anémies car une approche sélective du recueil des évènements indésirables a été utilisée
*** Une approche sélective du recueil des évènements indésirables a été utilisée
# Dans l’étude VOYAGER PAD
Tableau résumant les effets indésirables
Les fréquences des effets indésirables rapportés avec Xarelto dans la population adulte et pédiatrique sont résumées dans le Tableau 3 ci-dessous par classe de systèmes ou d’organes (classification MedDRA) et par fréquence.
Les fréquences sont définies comme suit :
très fréquent (≥ 1/10)
fréquent (≥ 1/100, <1/10)
peu fréquent (≥ 1/1 000, < 1/100)
rare (≥ 1/10 000, < 1/1000)
très rare (< 1/10000)
fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Tableau 3 : Ensemble des effets indésirables reportés chez les patients adultes dans les essais cliniques de phase III ou par une utilisation post-commercialisation* ainsi que dans deux études pédiatriques de phase II et deux études pédiatriques de phase III
Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéterminée |
Affections hématologiques et du système lymphatique | ||||
Anémie (dont résultat d’analyse de laboratoire correspondant) | Thrombocytose (dont élévation de la numération plaquettaire)A |
|
|
|
Affections du système immunitaire | ||||
| Réaction allergique, dermatite allergique |
| Réactions anaphylactiques, y compris choc anaphylactique |
|
Affections du système nerveux | ||||
Sensations vertigineuses, céphalées | Hémorragie cérébrale et intracrânienne, |
|
|
|
Affections oculaires | ||||
Hémorragie oculaire (dont hémorragie conjonctivale) |
|
|
|
|
Affections cardiaques | ||||
| Tachycardie |
|
|
|
Affections vasculaires | ||||
Hypotension, hématomes |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales | ||||
Épistaxis, hémoptysie |
|
| Pneumonie à éosinophiles |
|
Affections gastro-intestinales | ||||
Gingivorragie, hémorragie du tractus gastro-intestinal (dont rectorragie), douleurs gastro-intestinales et abdominales, dyspepsie, nausées, constipationA, diarrhée, vomissementsA | Sécheresse buccale |
|
|
|
Affections hépatobiliaires | ||||
Elévation des transaminases | Insuffisance hépatique, | Ictère, Elévation de la bilirubine conjuguée (avec ou sans élévation concomitante des ALAT), Cholestase, Hépatite (dont lésion hépatocellulaire) |
|
|
Affections de la peau et du tissu sous-cutané | ||||
Prurit (dont cas peu fréquents de prurit généralisé), éruption cutanée, ecchymose, hémorragie cutanée et sous-cutanée | Urticaire |
| Syndrome de Stevens-Johnson /Nécrolyse épidermique toxique, Syndrome DRESS |
|
Affections musculo-squelettiques et systémiques | ||||
Douleur des extrémitésA | Hémarthrose | Hémorragie musculaire |
| Syndrome de compression des loges secondaire à un saignement |
Affections du rein et des voies urinaires | ||||
Hémorragie du tractus urogénital (dont hématurie et ménorragieB), insuffisance rénale (dont élévation de la créatinine plasmatique, élévation de l’urée plasmatique) |
|
|
| Insuffisance rénale/insuffisance rénale aiguë secondaire à un saignement suffisant pour provoquer une hypoperfusion |
Troubles généraux et anomalies au site d’administration | ||||
FièvreA, | Sensation d’inconfort (dont malaise) | Oedème localiséA |
|
|
Investigations | ||||
| Élévation de la LDHA, de la lipaseA, de l’amylaseA |
|
|
|
Lésions, intoxications et complications liées aux procédures | ||||
Hémorragie post-opératoire (dont anémie postopératoire et hémorragie de la plaie), contusion, plaie suintanteA |
| Pseudoanévrisme vasculaireC |
|
|
A : effets observés dans la prévention des ETEV chez les patients adultes bénéficiant d’une intervention chirurgicale programmée de la hanche ou du genou (prothèse totale de hanche ou du genou)
B : effets observés très fréquemment chez les femmes âgées de < 55 ans dans le traitement de la TVP, de l’EP et la prévention des récidives
C : effets observés peu fréquemment dans la prévention des événements athérothrombotiques suite à un SCA (suite à une intervention coronaire percutanée)
* Une approche sélective prédéfinie du recueil des évènements indésirables a été utilisée dans certaines études de phase III. L’incidence des effets indésirables n’a pas augmenté et aucun nouvel effet indésirable médicamenteux n’a été identifié à la suite de l’analyse de ces études.
Description de certains effets indésirables
En raison du mode d’action pharmacologique du produit, l’utilisation de Xarelto peut être associée à un risque accru de saignement occulte ou apparent au niveau de tout organe ou tissu, ce qui peut entraîner une anémie post-hémorragique. Les signes, les symptômes et la sévérité (y compris les évolutions fatales) dépendront de la localisation et du degré ou de l’étendue du saignement et/ou de l’anémie (voir rubrique 4.9 « Prise en charge des saignements »). Au cours des études cliniques, des saignements des muqueuses (c. -à-d. épistaxis, saignement gingival, gastro-intestinal, génito-urinaire, dont des saignements vaginaux anormaux ou une augmentation des saignements menstruels) et des anémies ont été observés de manière plus fréquente durant le traitement au long cours par Xarelto comparé au traitement par AVK. Si nécessaire, des dosages de l’hémoglobine/des mesures de l’hématocrite pourraient permettre de détecter un saignement occulte et d’évaluer la pertinence clinique d’un saignement manifeste, en complément d’une surveillance clinique appropriée.
Le risque de saignement peut être augmenté chez certains groupes de patients, par ex. en cas d’hypertension artérielle sévère non contrôlée et/ou de traitement concomitant modifiant l’hémostase (voir rubrique 4.4 « Risque hémorragique »). Les saignements menstruels peuvent être amplifiés et/ou prolongés. Des complications hémorragiques peuvent se manifester sous forme de faiblesse, de pâleur, de sensations vertigineuses, de céphalées ou de gonflements inexpliqués, de dyspnée et d’état de choc inexpliqué. Dans certains cas, en conséquence de l’anémie, des symptômes d’ischémie cardiaque tels qu’une douleur thoracique ou une angine de poitrine, ont été observés.
Des complications connues, secondaires à une hémorragie sévère, telles qu’un syndrome de compression des loges et une insuffisance rénale due à l’hypoperfusion, ou une néphropathie liée aux anticoagulants ont été signalées sous Xarelto. Par conséquent, l’éventualité d’une hémorragie doit être envisagée lors de l’évaluation de toute affection chez un patient sous anticoagulant.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance


7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Bayer AG
51368 Leverkusen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHE
EU/1/08/472/025-035, EU/1/08/472/041, EU/1/08/472/046-047
10. DATE DE MISE À JOUR DU TEXTE
06/2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3680162 | XARELTO 10 MG COMP PELL 98 X 10 MG | B01AF01 | € 181,6 | - | Oui | - | - |
| 3569134 | XARELTO 2,5MG COMP PELL 56 X 2,5MG | B01AF01 | € 29,36 | - | Oui | € 7,57 | € 4,5 |
| 3786399 | XARELTO 2,5MG COMP PELL 196 X 2,5MG | B01AF01 | € 82,95 | - | Oui | € 15,9 | € 10,5 |