1. DÉNOMINATION DU MÉDICAMENT
Tremfya 100 mg OnePress solution injectable en stylo prérempli
Tremfya 100 mg PushPen solution injectable en stylo prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Tremfya 100 mg OnePress solution injectable en stylo prérempli
Chaque stylo prérempli contient 100 mg de guselkumab dans 1 mL de solution.
Tremfya 100 mg PushPen solution injectable en stylo prérempli
Chaque stylo prérempli contient 100 mg de guselkumab dans 1 mL de solution.
Le guselkumab est un anticorps monoclonal (AcMo) entièrement humain, de type immunoglobuline G1 lambda (IgG1λ) produit par des cellules ovariennes de hamster chinois (CHO) par la technologie de l’ADN recombinant.
Excipient(s) à effet notoire
Ce médicament contient 0,5 mg de polysorbate 80 (E433) dans chaque stylo prérempli, ce qui équivaut à 0,5 mg/mL.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable
La solution est limpide et incolore à jaune clair et peut contenir quelques petites particules blanches ou transparentes, avec un pH cible de 5,8 et une osmolarité d’environ 367,5 mOsm/L.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
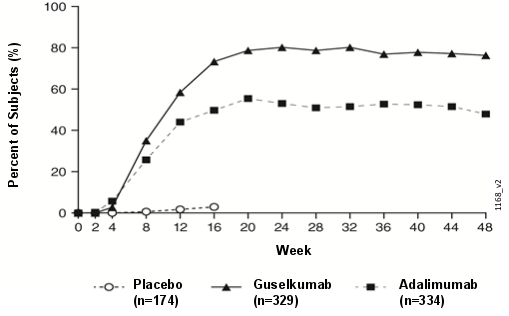
Psoriasis en plaques de l’adulte
Tremfya est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez l’adulte qui nécessite un traitement systémique.
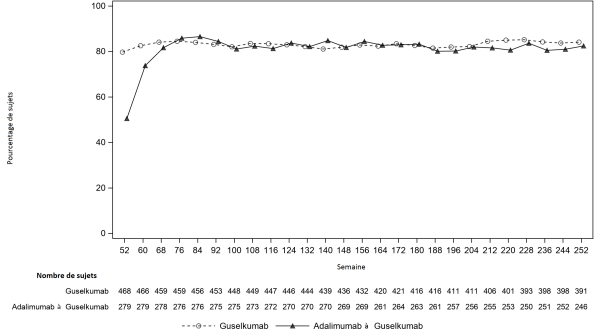
Rhumatisme psoriasique
Tremfya, seul ou en association avec le méthotrexate (MTX), est indiqué dans le traitement du rhumatisme psoriasique actif chez les patients adultes ayant présenté une réponse inadéquate ou une intolérance à un traitement de fond antirhumatismal (DMARD) antérieur (voir rubrique 5.1).
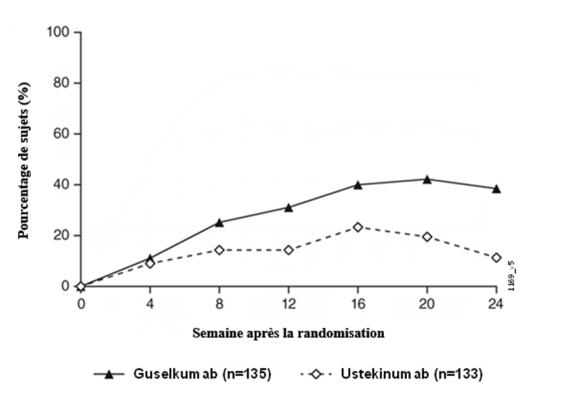
Rectocolite hémorragique
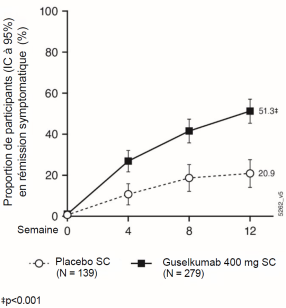
Tremfya est indiqué dans le traitement des patients adultes atteints de rectocolite hémorragique active modérée à sévère ayant présenté une réponse inadéquate, une perte de réponse ou une intolérance à un traitement conventionnel ou à un traitement biologique.
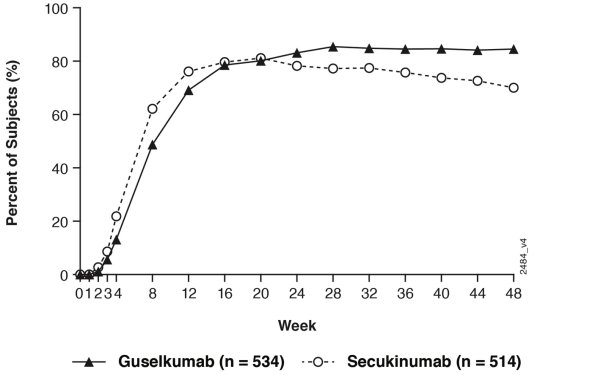
Maladie de Crohn
Tremfya est indiqué dans le traitement des patients adultes atteints de maladie de Crohn active modérée à sévère ayant présenté une réponse inadéquate, une perte de réponse ou une intolérance à un traitement conventionnel ou à un traitement biologique.
4.2 Posologie et mode d'administration
Ce médicament est destiné à être utilisé sous la conduite et la surveillance d’un médecin expérimenté dans le diagnostic et le traitement des pathologies pour lesquelles il est indiqué.
Posologie
Psoriasis en plaques
La dose recommandée est de 100 mg en injection sous-cutanée aux semaines 0 et 4, suivie d’une dose d’entretien toutes les 8 semaines.
L’arrêt du traitement doit être envisagé chez les patients ne présentant pas de réponse au bout de 16 semaines de traitement.
Rhumatisme psoriasique
La dose recommandée est de 100 mg en injection sous-cutanée aux semaines 0 et 4, suivie d’une dose d’entretien toutes les 8 semaines. Pour les patients présentant un risque élevé de lésion articulaire selon l’avis clinique, une dose de 100 mg toutes les 4 semaines peut être envisagée (voir rubrique 5.1).
L’arrêt du traitement doit être envisagé chez les patients ne présentant pas de réponse au bout de 24 semaines de traitement.
Rectocolite hémorragique
L’un des deux schémas posologiques d’induction suivants est recommandé :
- 200 mg administrés par perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8. Voir le RCP de Tremfya 200 mg, solution à diluer pour perfusion.
ou
- 400 mg administrés par injection sous-cutanée (administrés en deux injections consécutives de 200 mg chacune) à la semaine 0, à la semaine 4 et à la semaine 8. Voir le RCP de Tremfya 200 mg, solution injectable.
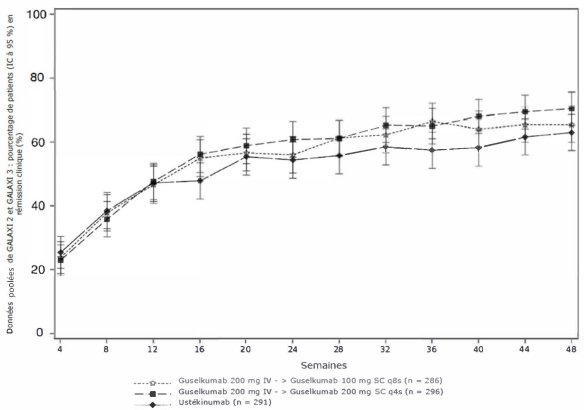
Suite au schéma posologique d’induction, la dose d’entretien recommandée à partir de la semaine 16 est de 100 mg administrée par injection sous-cutanée toutes les 8 semaines. En revanche, pour les patients qui, selon l’avis clinique, ne présentent pas de bénéfice thérapeutique adéquat au traitement d’induction, une dose d’entretien de 200 mg administrée par injection sous-cutanée à partir de la Semaine 12 puis toutes les 4 semaines peut être envisagée (voir rubrique 5.1). Pour la dose de 200 mg, voir le RCP de Tremfya 200 mg, solution injectable.
Les immunomodulateurs et/ou les corticoïdes peuvent être poursuivis pendant le traitement par guselkumab. Chez les patients ayant répondu au traitement par guselkumab, les corticoïdes peuvent être réduits ou arrêtés conformément à la pratique clinique.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 24 semaines de traitement.
Maladie de Crohn
L’un des deux schémas posologiques d’induction suivants est recommandé :
- 200 mg administrés par perfusion intraveineuse à la semaine 0, à la semaine 4 et à la semaine 8. Voir le RCP de Tremfya 200 mg, solution à diluer pour perfusion.
ou
- 400 mg administrés par injection sous-cutanée (administrés en deux injections consécutives de 200 mg chacune) à la semaine 0, à la semaine 4 et à la semaine 8. Voir le RCP de Tremfya 200 mg, solution injectable.
Suite au schéma posologique d’induction, la dose d’entretien recommandée à partir de la semaine 16 est de 100 mg administrée par injection sous-cutanée toutes les 8 semaines. En revanche, pour les patients qui, selon l’avis clinique, ne présentent pas de bénéfice thérapeutique adéquat au traitement d’induction, une dose d’entretien de 200 mg administrée par injection sous-cutanée à partir de la semaine 12 puis toutes les 4 semaines peut être envisagée (voir rubrique 5.1). Pour la dose de 200 mg, voir le RCP de Tremfya 200 mg, solution injectable.
Les immunomodulateurs et/ou les corticoïdes peuvent être poursuivis pendant le traitement par guselkumab. Chez les patients ayant répondu au traitement par guselkumab, les corticoïdes peuvent être réduits ou arrêtés conformément à la pratique clinique.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 24 semaines de traitement.
Dose oubliée
En cas d’oubli d’une dose, celle-ci doit être administrée dès que possible. Par la suite, la dose doit être reprise à la fréquence habituelle.
Populations particulières
Personnes âgées
Aucun ajustement posologique n'est nécessaire (voir rubrique 5.2).
Les données chez les patients âgés de 65 ans et plus sont limitées, et elles sont très limitées chez les patients âgés de 75 ans et plus (voir rubrique 5.2).
Insuffisance rénale ou hépatique
Tremfya n’a pas été étudié chez ces populations de patients. Ces pathologies n’ont généralement pas d’impact significatif sur la pharmacocinétique des anticorps monoclonaux, et aucun ajustement posologique n’est considéré comme nécessaire. Pour plus d’informations sur l’élimination du guselkumab, voir rubrique 5.2.
Population pédiatrique
La sécurité et l’efficacité de Tremfya chez les patients de moins de 18 ans atteints de rectocolite hémorragique, de maladie de Crohn et de rhumatisme psoriasique ainsi que chez les patients de moins de 6 ans atteints de psoriasis n’ont pas été établies. Aucune donnée n’est disponible. Tremfya 100 mg, solution injectable en stylo prérempli, n’est pas recommandé chez les enfants de moins de 18 ans en raison de données insuffisantes sur la sécurité et l’efficacité. Les données actuellement disponibles avec d’autres présentations sont décrites dans les rubriques 4.8, 5.1 et 5.2.
Mode d’administration
Par voie sous-cutanée uniquement. Les sites d’injection comprennent l’abdomen, la cuisse et l’arrière du haut du bras. Tremfya ne doit pas être injecté dans des zones où la peau présente une sensibilité, un bleu, une rougeur, un durcissement, un épaississement ou une desquamation. Dans la mesure du possible, les sites où la peau présente du psoriasis ne doivent pas être utilisés comme sites d’injection.
Après une formation adaptée à la technique d’injection sous-cutanée, les patients peuvent s’injecter Tremfya si le médecin estime cela approprié. Cependant, le médecin doit assurer un suivi médical adéquat des patients. Les patients doivent être informés de la nécessité d’injecter la dose complète de solution conformément aux « Instructions d’utilisation » fournies dans la boîte.
Pour les instructions concernant la préparation du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité grave à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Infection active et cliniquement importante (par exemple, tuberculose active ; voir rubrique 4.4).
4.8 Effets indésirables
Résumé du profil de sécurité
L’effet indésirable le plus fréquent était les infections des voies respiratoires (chez environ 8 % des patients dans les études sur la rectocolite hémorragique, 11 % des patients dans les études sur la maladie de Crohn, et 15 % des patients dans les études cliniques sur le psoriasis et le rhumatisme psoriasique).
Le profil de sécurité global chez les patients traités par Tremfya est similaire chez les patients atteints de psoriasis, de rhumatisme psoriasique, de rectocolite hémorragique, et de maladie de Crohn.
Tableau récapitulatif des effets indésirables
Le tableau 1 fournit une liste des effets indésirables observés dans les études cliniques sur le psoriasis, le rhumatisme psoriasique, la rectocolite hémorragique, et la maladie de Crohn, ainsi que des effets indésirables rapportés depuis la mise sur le marché du produit. Les effets indésirables sont présentés par classe de système d’organes MedDRA et par fréquence, selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 1: Liste des effets indésirables | ||
Classe de système d’organes | Fréquence | Effets indésirables |
Infections et infestations | Très fréquent | Infections des voies respiratoires |
Peu fréquent | Infections à Herpes simplex | |
Peu fréquent | Dermatophytoses | |
Peu fréquent | Gastro-entérite | |
Affections du système immunitaire | Rare | Hypersensibilité |
Rare | Anaphylaxie | |
Affections du système nerveux | Fréquent | Céphalée |
Affections gastro-intestinales | Fréquent | Diarrhée |
Affections de la peau et du tissus sous-cutané | Fréquent | Eruption cutanée |
Peu fréquent | Urticaire | |
Affections musculo-squelettiques et systémiques | Fréquent | Arthralgie |
Troubles généraux et anomalies au site d’administration | Fréquent | Réactions au site d’injection |
Investigations | Fréquent | Transaminases augmentées |
Peu fréquent | Neutrophiles diminués | |
Description de certains effets indésirables
Transaminases augmentées
Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le rhumatisme psoriasique, les effets indésirables de type augmentation des transaminases (comprenant ALAT augmentée, ASAT augmentée, enzymes hépatiques augmentées, transaminases augmentées, test hépatique anormal, hypertransaminasémie) ont été rapportés plus fréquemment dans les groupes traités par guselkumab (8,6 % dans le groupe recevant 100 mg par voie sous-cutanée toutes les 4 semaines et 8,3 % dans le groupe recevant 100 mg par voie sous-cutanée toutes les 8 semaines) que dans le groupe placebo (4,6 %). En un an, les effets indésirables de type augmentation des transaminases (ci-dessus) ont été rapportés chez 12,9 % des patients dans le groupe toutes les 4 semaines et 11,7 % des patients dans le groupe toutes les 8 semaines.
Sur la base des analyses biologiques, la plupart des augmentations des transaminases (ALAT et ASAT) étaient ≤ 3 x la limite supérieure de la normale (LSN). Les augmentations des transaminases situées entre > 3 et ≤ 5 x LSN et > 5 x LSN étaient peu fréquentes, survenant plus souvent dans le groupe guselkumab toutes les 4 semaines que dans le groupe guselkumab toutes les 8 semaines (tableau 2). Une fréquence similaire a été observée quels que soit la sévérité et le bras de traitement à la fin de l’étude clinique de phase III de 2 ans sur le rhumatisme psoriasique.
Tableau 2 : Fréquence de patients présentant une augmentation des transaminases post-inclusion dans les études cliniques de phase III sur le rhumatisme psoriasique | |||||
| Jusqu’à la semaine 24a | Jusqu’à 1 anb | |||
| Placebo | guselkumab | guselkumab | guselkumab | guselkumab |
ALAT | |||||
> 1 à ≤3 x LSN | 30,0% | 28,2% | 35,0% | 33,5% | 41,2% |
> 3 à ≤5 x LSN | 1,4% | 1,1% | 2,7% | 1,6% | 4,6% |
> 5 x LSN | 0,8% | 0,8% | 1,1% | 1,1% | 1,1% |
ASAT | |||||
> 1 à ≤3 x LSN | 20,0% | 18,8% | 21,6% | 22,8% | 27,8% |
> 3 à ≤5 x LSN | 0,5% | 1,6% | 1,6% | 2,9% | 3,8% |
> 5 x LSN | 1,1% | 0,5% | 1,6% | 0,5% | 1,6% |
a période contrôlée versus placebo | |||||
Dans les études cliniques sur le psoriasis, avec une dose de guselkumab toutes les 8 semaines, la fréquence des augmentations des transaminases (ALAT et ASAT), évaluée sur une période d’un an, a été similaire à celle observée dans les études cliniques sur le rhumatisme psoriasique avec une dose de guselkumab toutes les 8 semaines. Sur une période de 5 ans, l’incidence de l’augmentation des transaminases n’a pas augmenté par année de traitement sous guselkumab. La plupart des augmentations de transaminase étaient ≤ 3 x LSN.
Dans la plupart des cas, l’augmentation des transaminases était transitoire et n’a pas entraîné l’arrêt du traitement.
Dans les études cliniques poolées de Phase II et de Phase III sur la maladie de Crohn, au cours de la période d’induction contrôlée par placebo (semaines 0 à 12), des effets indésirables de type augmentation des transaminases (comprenant ALAT augmentée, ASAT augmentée, enzyme hépatique augmentée, transaminases augmentées, et test de la fonction hépatique augmenté) ont été rapportés plus fréquemment dans les groupes traités par guselkumab (1,7 % des patients) que dans le groupe placebo (0,6 % des patients). Dans les études cliniques poolées de phase II et de phase III sur la maladie de Crohn, pendant la période étudiée d’environ un an, des effets indésirables de type augmentation des transaminases (comprenant ALAT augmentée, ASAT augmentée, enzyme hépatique augmentée, transaminases augmentées, fonction hépatique anormal, et test de la fonction hépatique augmenté) ont été rapportés chez 3,4 % des patients dans le groupe traité par guselkumab 200 mg par voie sous-cutanée toutes les 4 semaines et 4,1 % des patients dans le groupe traité par guselkumab 100 mg par voie sous-cutanée toutes les 8 semaines, contre 2,4 % des patients dans le groupe placebo.
Sur la base des analyses biologiques des études cliniques poolées de Phase II et de Phase III sur la maladie de Crohn, la fréquence d’augmentation d’ALAT ou d’ASAT était inférieure à celle observée dans les études cliniques de phase III sur le rhumatisme psoriasique. Dans les études cliniques poolées de phase II et de phase III sur la maladie de Crohn, pendant la période contrôlée par placebo (semaine 12), des augmentations d’ALAT (< 1 % des patients) et d’ASAT (< 1 % des patients) ≥ 3 x LSN ont été rapportées chez des patients traités par guselkumab. Dans les études cliniques poolées de phase II et de phase III sur la maladie de Crohn, pendant la période étudiée d’environ un an, des augmentations d’ALAT et/ou d’ASAT ≥ 3 x LSN ont été rapportées chez 2,7 % des patients dans le groupe traité par guselkumab 200 mg par voie sous-cutanée toutes les 4 semaines et chez 2,6 % des patients dans le groupe traité par guselkumab 100 mg par voie sous-cutanée toutes les 8 semaines, contre 1,9 % dans le groupe placebo. Dans la plupart des cas, l’augmentation des transaminases était transitoire et n’a pas entraîné l’arrêt du traitement.
Diminution du nombre de neutrophiles
Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le rhumatisme psoriasique, l’effet indésirable de type diminution du nombre de neutrophiles a été rapporté plus fréquemment dans le groupe traité par guselkumab (0,9 %) que dans le groupe placebo (0 %). En un an, l’effet indésirable de type diminution du nombre de neutrophiles a été rapporté chez 0,9 % des patients traités par guselkumab. Dans la plupart des cas, la diminution du nombre de neutrophiles sanguins a été légère, transitoire, non associée à une infection et n’a pas entraîné d’arrêt du traitement.
Gastro-entérite
Pendant la période contrôlée versus placebo de deux études cliniques de Phase III sur le psoriasis, des gastro-entérites sont survenues plus fréquemment dans le groupe traité par guselkumab (1,1 %) que dans le groupe placebo (0,7 %). Jusqu’à la semaine 264, 5,8 % de tous les patients traités par guselkumab ont rapporté une gastro-entérite. Ces gastro-entérites étaient non graves et n’ont pas conduit à l’arrêt du traitement par guselkumab jusqu’à la semaine 264. Les taux de gastro-entérite observés pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique étaient similaires à ceux observés dans les études cliniques sur le psoriasis.
Réactions au site d’injection
Lors de deux études cliniques de Phase III sur le psoriasis, 0,7 % des injections de guselkumab et 0,3 % des injections de placebo ont été associées à des réactions au site d’injection jusqu’à la semaine 48. Jusqu’à la semaine 264, 0,4 % des injections de guselkumab ont été associées à des réactions au site d’injection. Ces réactions au site d’injection étaient généralement de sévérité légère à modérée ; aucune n’était grave, et une seule a conduit à l’arrêt du traitement par guselkumab.
Lors de deux études cliniques de Phase III sur le rhumatisme psoriasique jusqu’à la semaine 24, le nombre de patients pour lesquels une ou plusieurs réactions au site d’injection ont été rapportées était faible et légèrement plus élevé dans les groupes guselkumab que dans le groupe placebo ; 5 patients (1,3 %) dans le groupe guselkumab toutes les 8 semaines, 4 patients (1,1 %) dans le groupe guselkumab toutes les 4 semaines et 1 patient (0,3 %) dans le groupe placebo. Un patient a arrêté le guselkumab en raison d’une réaction au site d’injection pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique. En un an, la proportion de patients ayant présenté 1 réaction au site d’injection ou plus était de 1,6 % et de 2,4 % dans les groupes guselkumab toutes les 8 semaines et toutes les 4 semaines, respectivement. Dans l’ensemble, le taux d’injections associées à des réactions au site d’injection observé pendant la période contrôlée versus placebo des études cliniques sur le rhumatisme psoriasique était similaire aux taux observés dans les études cliniques sur le psoriasis.
Dans l’étude clinique de Phase III d’entretien dans la rectocolite hémorragique jusqu’à la semaine 44, la proportion de patients ayant rapporté une ou plusieurs réactions au site d’injection du guselkumab était de 7,9 % (2,5 % des injections) dans le groupe guselkumab 200 mg par voie sous-cutanée toutes les 4 semaines (le guselkumab 200 mg était administré sous forme de deux injections de 100 mg dans l’étude clinique de phase III d’entretien dans la rectocolite hémorragique) et aucune réaction au site d’injection dans le groupe guselkumab 100 mg par voie sous-cutanée toutes les 8 semaines. La plupart des réactions au site d’injection étaient légères et aucune n’était grave.
Dans les études cliniques de Phase II et de Phase III sur la maladie de Crohn jusqu’à la semaine 48, la proportion de patients ayant rapporté une ou plusieurs réactions au site d’injection du guselkumab était de 4,1 % (0,8 % des injections) dans le groupe traité par 200 mg de guselkumab en induction intraveineuse, suivi de 200 mg par voie sous-cutanée toutes les 4 semaines, et de 1,4 % (0,6 % des injections) dans le groupe guselkumab 200 mg en induction intraveineuse, suivi de 100 mg par voie sous-cutanée toutes les 8 semaines. Globalement, les réactions au site d’injection étaient légères et aucune n’était grave.
Dans une étude clinique de Phase III sur la maladie de Crohn jusqu’à la semaine 48, la proportion de patients ayant rapporté une ou plusieurs réactions au site d’injection du guselkumab était de 7 % (1,3 % des injections) dans le groupe traité par 400 mg de guselkumab en induction sous-cutanée suivi de 200 mg par voie sous-cutanée toutes les 4 semaines et de 4,3 % (0,7 % des injections) dans le groupe guselkumab 400 mg en induction sous-cutanée suivi de 100 mg par voie sous-cutanée toutes les 8 semaines. La plupart des réactions au site d’injection étaient légères et aucune n’était grave.
Immunogénicité
L’immunogénicité du guselkumab a été évaluée à l’aide d’une méthode sensible de dosage immunologique, tolérante au biomédicament.
D’après les analyses des études poolées de Phase II et de Phase III menées auprès de patients atteints de psoriasis et de rhumatisme psoriasique, 5 % (n = 145) des patients traités par guselkumab ont développé des anticorps anti-médicament sur une durée de traitement allant jusqu’à 52 semaines. Parmi les patients ayant développé des anticorps anti-médicament, environ 8 % (n = 12) présentaient des anticorps catégorisés comme neutralisants, soit 0,4 % de l’ensemble des patients traités par guselkumab. Dans les analyses poolées de Phase III, parmi les patients atteints de psoriasis, environ 15 % des patients traités par guselkumab ont développé des anticorps anti-médicament sur une durée de traitement allant jusqu’à 264 semaines. Parmi les patients ayant développé des anticorps anti-médicament, environ 5 % présentaient des anticorps qualifiés de neutralisants, soit 0,76 % de l’ensemble des patients traités par guselkumab. La présence d’anticorps anti-médicament n’a pas été associée à une réduction de l’efficacité ou à la survenue de réactions au site d’injection.
Dans les analyses poolées de Phase II et de Phase III réalisées chez des patients atteints de rectocolite hémorragique traités par induction intraveineuse suivie d’un traitement d’entretien sous-cutané, environ 12 % (n = 58) des patients traités par guselkumab pendant une période allant jusqu'à 56 semaines ont développé des anticorps anti-médicament. Parmi les patients ayant développé des anticorps anti-médicaments, environ 16 % (n = 9) présentaient des anticorps qualifiés de neutralisants, soit 2 % de l’ensemble des patients traités par guselkumab. Dans une analyse de Phase III jusqu’à la semaine 24 chez des patients atteints de rectocolite hémorragique traités par induction sous-cutanée suivi d’un traitement d’entretien sous-cutané, environ 9 % (n = 24) des patients traités par guselkumab ont développé des anticorps anti-médicament. Parmi les patients ayant développé des anticorps anti-médicament, 13 % (n = 3) présentaient des anticorps qualifiés de neutralisants, soit 1 % de l’ensemble des patients traités par guselkumab. La présence d’anticorps anti-médicament n'a pas été associée à une réduction de l’efficacité ou à la survenue de réactions au site d'injection.
Dans les analyses poolées de Phase II et de Phase III jusqu’à la semaine 48 chez les patients atteints de la maladie de Crohn traités par induction intraveineuse suivie d’un schéma posologique d’entretien sous-cutané, environ 5 % (n = 30) des patients traités par guselkumab ont développé des anticorps anti-médicament. Parmi les patients ayant développé des anticorps anti-médicament, environ 7 % (n = 2) présentaient des anticorps qualifiés de neutralisants, soit 0,3 % de l’ensemble des patients traités par guselkumab. Dans une analyse de Phase III jusqu’à la semaine 48 chez des patients atteints de la maladie de Crohn traités par induction sous-cutanée suivi d’un schéma posologique d’entretien sous-cutané, environ 9 % (n = 24) des patients traités par guselkumab ont développé des anticorps anti-médicament. Parmi ces patients, 13 % (n = 3) présentaient des anticorps qualifiés de neutralisants, soit 1 % de l’ensemble des patients traités par guselkumab. La présence d’anticorps anti-médicament n’a pas été associée à une réduction de l’efficacité ou à la survenue de réactions au site d’injection.
Population pédiatrique
Psoriasis en plaques
La sécurité du guselkumab a été évalué dans le cadre d’une étude de Phase III contrôlée versus placebo et traitement actif menée chez des patients pédiatriques atteints de psoriasis en plaques modéré à sévère. Cette étude clinique a évalué la sécurité pendant une période pouvant aller jusqu’à 52 semaines chez 120 patients âgés de 6 à 17 ans. Le profil de sécurité de l’injection sous-cutanée de guselkumab utilisant le stylo prérempli de 45 mg/0,45 mL ou la seringue préremplie de 100 mg chez les patients pédiatriques âgés de 6 à 17 ans présentait un profil de sécurité cohérent avec celui rapporté dans les études sur le psoriasis en plaques chez l’adulte (voir rubrique 4.2).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance
Site internet : www.notifieruneffetindesirable.be
E-mail : adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgique
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
Tremfya 100 mg, OnePress solution injectable en stylo prérempli
EU/1/17/1234/002 1 stylo prérempli
EU/1/17/1234/003 2 stylos préremplis
Tremfya 100 mg PushPen solution injectable en stylo prérempli
EU/1/17/1234/010 1 stylo prérempli
EU/1/17/1234/011 2 stylos préremplis
10. DATE DE MISE À JOUR DU TEXTE
18/12/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3782646 | TREMFYA 100MG ONEPRESS SOL INJ STYLO PREREMPLI 1 | L04AC16 | € 1989,38 | - | Oui | € 12,8 | € 8,5 |