RESUME DES CARACTERISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Bydureon 2 mg, suspension injectable à libération prolongée en stylo prérempli.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque stylo prérempli délivre une dose de 2 mg d’exénatide dans 0,85 mL.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Suspension injectable à libération prolongée en stylo prérempli (BCise).
Suspension opaque de couleur blanche à blanc cassé.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Bydureon est indiqué chez les adultes, les adolescents et les enfants âgés de 10 ans et plus atteints de diabète de type 2 pour améliorer le contrôle de la glycémie en association avec d’autres médicaments destinés au traitement du diabète incluant l’insuline basale, lorsque le traitement en cours, en complément d’un régime alimentaire adapté et d’une activité physique, ne permet pas d’assurer un contrôle adéquat de la glycémie.
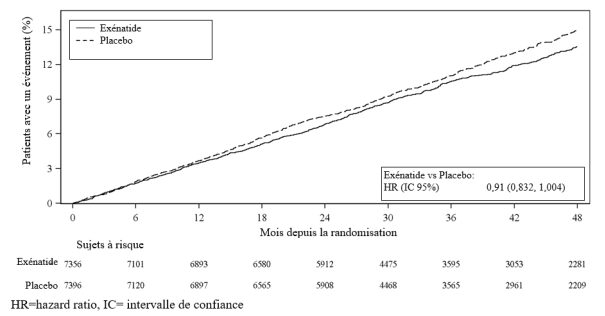
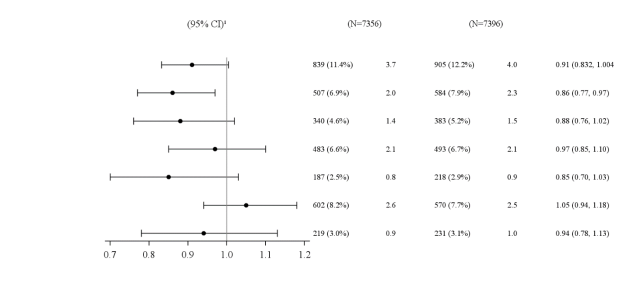
Pour les résultats des études concernant les associations, les effets sur le contrôle glycémique et les événements cardiovasculaires, et les populations étudiées, voir les rubriques 4.4, 4.5 et 5.1.
4.2 Posologie et mode d’administration
Posologie
La dose recommandée est de 2 mg d’exénatide une fois par semaine.
Chez les patients passant du traitement par l’exénatide à libération immédiate (Byetta) à l’exénatide à libération prolongée (Bydureon ou Bydureon BCise), il peut être observé des augmentations transitoires de la glycémie. La situation s’améliore généralement dans les quatre premières semaines qui suivent l’initiation du traitement. Il est possible de passer d’un produit à base d’exénatide à libération prolongée à l’autre (Bydureon ou Bydureon BCise), sans qu’aucun effet significatif sur la glycémie ne soit attendu.
Quand l’exénatide à libération prolongée est associé à un traitement par metformine et/ou une thiazolidinedione, le traitement par metformine et/ou une thiazolidinedione peut être poursuivi à la même posologie. Quand il est associé à un traitement par un sulfamide hypoglycémiant, une diminution de la posologie du sulfamide hypoglycémiant doit être envisagée afin de diminuer le risque d’hypoglycémie (voir rubrique 4.4). L’association avec la thiazolidinedione n'a été étudiée que chez les patients adultes.
L’exénatide à libération prolongée doit être administré une fois par semaine, le même jour chaque semaine. Le jour de l’administration hebdomadaire peut être modifié si nécessaire à condition que la dernière dose ait été administrée au moins trois jours avant. L’exénatide à libération prolongée peut être administré à n’importe quel moment de la journée, avec ou sans repas.
En cas d’oubli d’une dose, celle-ci doit être administrée dès que possible, à condition que la dose suivante soit prévue au moins 3 jours plus tard. Les patients peuvent ensuite reprendre l’administration hebdomadaire habituelle.
En cas d’oubli d’une dose et la dose suivante étant prévue 1 ou 2 jours plus tard, le patient ne doit pas s’administrer la dose oubliée et doit reprendre l’administration de l’exénatide à libération prolongée le jour suivant prévu.
L’utilisation de ce médicament ne nécessite pas d’autosurveillance supplémentaire. Une autosurveillance glycémique est nécessaire afin d’ajuster la dose des sulfamides hypoglycémiants ou d’insuline, notamment lors de l’instauration du traitement par exénatide à libération prolongée et de la réduction de l’insuline. L’adoption d’une approche par étapes de la réduction de la dose d’insuline est recommandée.
Si un traitement hypoglycémiant différent est initié après l’arrêt de l’exénatide à libération prolongée, la libération prolongée du produit doit être prise en compte (voir rubrique 5.2).
Populations particulières
Sujets âgés
Aucun ajustement posologique n’est nécessaire en fonction de l’âge. Cependant, la fonction rénale du patient doit être prise en compte, car elle diminue généralement avec l’âge (voir Atteinte de la fonction rénale) (voir rubrique 5.2).
Atteinte de la fonction rénale
Aucun ajustement posologique n’est nécessaire chez les patients avec une atteinte légère ou modérée de la fonction rénale.
L’exénatide à libération prolongée n’est pas recommandé chez les patients avec une pathologie rénale terminale ou une atteinte sévère de la fonction rénale (débit de filtration glomérulaire [DFG] < 30 mL/min) (voir rubrique 4.4).
Atteinte de la fonction hépatique
Aucun ajustement posologique n’est nécessaire chez les patients avec une atteinte de la fonction hépatique (voir rubrique 5.2).
Population pédiatrique
Aucun ajustement posologique n'est nécessaire pour les adolescents et les enfants âgés de 10 ans et plus. Aucune donnée n'est disponible pour les enfants âgés de moins de 10 ans (voir rubriques 5.1 et 5.2).
Mode d’administration
Voie sous-cutanée
L’exénatide à libération prolongée est à administrer par le patient lui-même. Chaque stylo ne peut être utilisé que par une personne et une seule fois.
Avant l’initiation de l’exénatide à libération prolongée, il est fortement recommandé que les patients et les soignants soient formés par leur professionnel de santé. Le « Manuel d’utilisation », fourni à l’intérieur de la boîte, doit être suivi attentivement.
Chaque dose doit être administrée par injection sous-cutanée dans l’abdomen, la cuisse, ou l’arrière du bras immédiatement après le mélange complet du médicament.
Quand il est associé à de l’insuline, l’exénatide à libération prolongée et l’insuline doivent être administrés en deux injections séparées.
Pour les instructions concernant la préparation du médicament avant administration, voir la rubrique 6.6 et le « Manuel d’utilisation ».
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité d’emploi
Les effets indésirables les plus fréquents chez les adultes au cours des études cliniques étaient gastro-intestinaux (principalement des nausées (8 %), qui ont eu tendance à se dissiper avec la poursuite du traitement), des céphalées (4 %) et des réactions au site d’injection, telles qu’un prurit (3 %) et un érythème (2 %). Par ailleurs, des cas d’hypoglycémie sont survenus très fréquemment avec un sulfamide hypoglycémiant (voir Description des effets indésirables sélectionnés, ci-dessous). La plupart des effets indésirables étaient d’intensité légère à modérée.
Tableau récapitulatif des effets indésirables
Les fréquences des effets indésirables de Bydureon BCise identifiés à partir des études cliniques chez les adultes sont résumées dans le Tableau 1 ci-dessous.
Les données poolées des études cliniques portant sur Bydureon BCise comprennent deux études de phase III contrôlées versus comparateur d’une durée de 6 à 12 mois chez les adultes. Les phases de suivi et d’extension des études sont incluses dans ces données poolées. Les traitements de base incluaient un régime alimentaire adapté et une activité physique seuls ou avec de la metformine, un sulfamide hypoglycémiant, une thiazolidinedione ou une association de traitements hypoglycémiants oraux. Les effets indésirables observés avec l’exénatide à libération prolongée en dehors des études cliniques portant sur Bydureon BCise sont également inclus dans le Tableau 1.
Les traitements de base dans les études cliniques de l’exénatide à libération prolongée incluaient un régime alimentaire adapté et une activité physique, la metformine, un sulfamide hypoglycémiant, une thiazolidinedione, une association de traitements hypoglycémiants oraux ou une insuline basale.
Les effets indésirables sont listés ci-dessous selon la terminologie MedDRA par classe de système d’organes et par fréquence absolue. Les fréquences sont définies de la manière suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 : Effets indésirables de Bydureon BCise identifiés à partir des études cliniques et de rapports spontanés chez les adultes
Classe de système d’organes/Effets indésirables | Fréquence de survenue1 | |||||
| Très fréquent | Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéterminée |
Affections hématologiques et du système lymphatique | ||||||
Thrombocytopénie d’origine médicamenteuse9 |
|
|
|
|
| X |
Affections hépatobiliaires |
|
|
|
|
|
|
Cholécystite11 |
|
| X |
|
|
|
Lithiase biliaire |
|
| X |
|
|
|
Affections du système immunitaire | ||||||
Réaction anaphylactique2 |
|
|
| X |
|
|
Troubles du métabolisme et de la nutrition | ||||||
Hypoglycémie (avec un sulfamide hypoglycémiant) 5,6.7 | X |
|
|
|
|
|
Hypoglycémie (sans sulfamide hypoglycémiant)5,6,7 |
|
| X |
|
|
|
Hypoglycémie (avec insuline)3,4,5 |
| X |
|
|
|
|
Diminution de l’appétit |
|
| X |
|
|
|
Déshydratation |
|
| X |
|
|
|
Affections du système nerveux | ||||||
Céphalées |
| X |
|
|
|
|
Sensation vertigineuse |
| X |
|
|
|
|
Dysgueusie |
|
| X |
|
|
|
Somnolence2 |
|
| X |
|
|
|
Affections gastro-intestinales | ||||||
Nausées5 |
| X |
|
|
|
|
Diarrhée |
| X |
|
|
|
|
Vomissements |
| X |
|
|
|
|
Constipation |
| X |
|
|
|
|
Dyspepsie |
| X |
|
|
|
|
Reflux gastro-œsophagien |
| X |
|
|
|
|
Distension abdominale |
| X |
|
|
|
|
Douleur abdominale |
| X |
|
|
|
|
Flatulences |
|
| X |
|
|
|
Pancréatite aiguë (voir rubrique 4.4) |
|
| X |
|
|
|
Éructation2 |
|
| X |
|
|
|
Occlusion intestinale2 |
|
| X |
|
|
|
Retard de la vidange gastrique10 |
|
| X |
|
|
|
Affections de la peau et du tissu sous-cutané | ||||||
Urticaire |
|
| X |
|
|
|
Hyperhidrose |
|
| X |
|
|
|
Éruption maculaire ou papuleuse |
|
| X |
|
|
|
Prurit |
|
| X |
|
|
|
Alopécie2 |
|
| X |
|
|
|
Angio-œdème9 |
|
|
|
|
| X |
Abcès et cellulite au site d’injection9 |
|
|
|
|
| X |
Affections du rein et des voies urinaires | ||||||
Altération de la fonction rénale8 |
|
| X |
|
|
|
Troubles généraux et anomalies au site d’administration | ||||||
Prurit au site d’injection5 |
| X |
|
|
|
|
Affections du rein et des voies urinaires | ||||||
Altération de la fonction rénale8 |
|
| X |
|
|
|
Troubles généraux et anomalies au site d’administration | ||||||
Prurit au site d’injection5 |
| X |
|
|
|
|
Érythème au site d’injection5 |
| X |
|
|
|
|
Fatigue |
| X |
|
|
|
|
Réaction au site d’injection5 |
|
| X |
|
|
|
Asthénie |
|
| X |
|
|
|
Éruption au site d’injection5 |
|
| X |
|
|
|
Sensation de nervosité2 |
|
|
| X |
|
|
Investigations | ||||||
|
|
|
|
|
|
|
INR augmenté9 (voir rubrique 4.4) |
|
|
|
|
| X |
1 Fréquence basée sur les études terminées d’efficacité et de sécurité à long terme (n = 526), sauf indication contraire. Comprend le suivi de soixante-dix jours suivant la dernière dose reçue et la période d’extension.
2 Fréquence basée sur douze études terminées d’efficacité et de sécurité à long terme de l’exénatide à libération prolongée. n = 2 868 au total.
3 Basée sur les événements d’hypoglycémie qui 1. ont entraîné une perte de connaissance, des convulsions ou un coma qui se sont résolus après administration de glucagon ou de glucose OU 2. demandent l’assistance d’une tierce personne pour la prise en charge du fait d’une altération de la conscience ou du comportement et sont associés à une glycémie < 54 mg/dL (3 mmol/L) OU 3. entraînent des symptômes d’une hypoglycémie avec une glycémie concomitante < 54 mg/dL (3 mmol/L) avant traitement.
4. Fréquence rapportée de la période de traitement contrôlée de 28 semaines de l’étude de l’exénatide à libération prolongée en ajout de l’insuline glargine (n = 231).
5 Voir Description des effets indésirables sélectionnés, ci-dessous.
6 Fréquences rapportées dans les données poolées des périodes contrôlées des deux études cliniques de phase III (n = 410).
7 Basée sur les événements d’hypoglycémie qui présentent des symptômes d’une hypoglycémie avec une glycémie concomitante < 54 mg/dL (3 mmol/L) avant traitement.
8 Inclut insuffisance rénale aiguë, aggravation d’une insuffisance rénale chronique, atteinte rénale, augmentation de la créatinine sérique. Voir rubrique 4.4.
9 Fréquence basée sur les données issues des notifications spontanées de l’exénatide à libération prolongée (dénominateur inconnu).
10 Fréquence établie à partir de 16 études d'efficacité et de sécurité à long terme réalisées avec l'exénatide à libération prolongée N = 4086 au total.
11 Fréquence établie à partir des études d'innocuité et d'efficacité réalisées avec BYDUREON (n=3560 au total) ; comprend les études DURATION 7 et DURATION 8.
Description des effets indésirables sélectionnés
Thrombocytopénie d’origine médicamenteuse
Une thrombocytopénie d’origine médicamenteuse, avec des anticorps anti-plaquettaires induits par l'exénatide, a été signalée chez les adultes dans le cadre du suivi post-commercialisation. La thrombocytopénie d’origine médicamenteuse est une réaction à médiation immunitaire qui est causée par des anticorps, médicament dépendant, ciblant les plaquettes. Ces anticorps entrainent une destruction des plaquettes en présence du médicament sensibilisant.
Hypoglycémie
Aucun événement d’hypoglycémie majeure n’a été observé avec Bydureon BCise dans les études cliniques menées chez les adultes. L’incidence globale des hypoglycémies mineures était de 6,3 %. Cette incidence était augmentée quand il était utilisé en association avec un sulfamide hypoglycémiant (26,1 %) versus sans sulfamide hypoglycémiant (0,9 %) (voir rubrique 4.4). Afin de réduire le risque d’hypoglycémie associé à l’utilisation d’un sulfamide hypoglycémiant, une réduction de la dose de sulfamide hypoglycémiant peut être envisagée (voir rubriques 4.2 et 4.4).
Quand l’exénatide à libération prolongée était ajouté à l’insuline basale, aucun ajustement de la dose initiale d’insuline ne s’est avéré nécessaire. L’exénatide à libération prolongée en association avec l’insuline basale n’a montré aucune différence cliniquement significative de l’incidence des épisodes d’hypoglycémie par rapport à l’insuline. Aucun épisode d’hypoglycémie majeure n’a été rapporté dans le groupe de l’exénatide à libération prolongée associé à l’insuline.
Nausées
L’effet indésirable gastro-intestinal rapporté le plus fréquemment chez les adultes était des nausées. Pendant la période contrôlée de l’étude clinique comparant Bydureon BCise à l’exénatide à libération immédiate, des nausées ont été rapportées chez respectivement 9,6 % et 20,5 % des patients de chaque groupe. Dans l’ensemble, 9,3 % des patients traités par Bydureon BCise ont rapporté des nausées pendant la période contrôlée des deux études cliniques. La plupart des épisodes de nausées étaient d’intensité légère à modérée, ils étaient associés à l’initiation du traitement et leur fréquence a diminué avec le temps.
Réactions au site d’injection
Pendant la période contrôlée des études cliniques menées chez les adultes, des réactions au site d’injection ont été observées plus fréquemment chez les patients traités par Bydureon BCise que chez ceux traités par le comparateur (24 % versus 4 % avec l’exénatide à libération immédiate). Ces réactions au site d’injection ont généralement été d’intensité légère et n’ont d’ordinaire pas conduit à l’arrêt du médicament à l’étude. Les patients peuvent recevoir un traitement symptomatique pour les soulager, tout en continuant le traitement. Un site d’injection différent doit être utilisé chaque semaine. En post-commercialisation avec l’exénatide à libération prolongée, des cas d’abcès et de cellulite au site d’injection ont été rapportés.
Des nodules sous-cutanés au site d’injection ont été observés fréquemment au cours des études cliniques, ce qui est cohérent avec les propriétés connues des formulations en microsphères de polymère poly (D,L-lactide-co-glycolide). La plupart de ces nodules n’influaient pas sur la participation à l’étude et disparaissaient avec le temps.
Immunogénicité
Compte tenu des propriétés potentiellement immunogènes des protéines et des peptides, les patients traités par l’exénatide à libération prolongée peuvent développer des anticorps anti-exénatide.
Approximativement 42 % des patients ont développé un faible taux d’anticorps anti-exénatide et 32 % des patients ont développé un taux élevé d’anticorps à tout moment au cours des études menées chez les adultes. Le pourcentage de patients avec anticorps, en particulier avec un taux élevé, était maximal environ 8 à 16 semaines après l’administration, puis a diminué avec le temps. À la fin de l’étude, approximativement 43 % des patients avaient un faible taux d’anticorps anti-exénatide et 14 % des patients avaient un taux élevé d’anticorps. Globalement, le contrôle glycémique (HbA1c) chez les patients traités par Bydureon BCise et présentant un faible taux d’anticorps à la dernière visite (-1,1 % à -1,5 %) était comparable à celui observé chez les patients sans anticorps (-1,1 % à -1,4 %). Même si les patients ayant un taux élevé d’anticorps à la dernière visite présentaient une réponse HbA1c atténuée, les réductions de l’HbA1c chez ces patients étaient cliniquement pertinentes (-0,6 % à ‑0,7 %).
Parmi les patients adultes traités par Bydureon BCise et évaluables pour les anticorps (n = 393), l’incidence des réactions au site d’injection potentiellement immunogènes (le plus souvent nodule au site d’injection) au cours des deux études était d’environ 20 %. Ces réactions étaient moins fréquemment observées chez les patients sans anticorps (16 %) et ceux ayant un faible taux d’anticorps (16 %) que chez les patients avec un taux d’anticorps élevé (27 %).
Perte de poids rapide
Dans une étude de 30 semaines menée chez les adultes, approximativement 3 % (n = 4/148) des patients traités par l’exénatide à libération prolongée ont présenté au moins une période de perte de poids rapide (perte de poids supérieure à 1,5 kg/semaine enregistrée entre deux visites d’étude consécutives).
Augmentation de la fréquence cardiaque
Une augmentation moyenne de la fréquence cardiaque (FC) de 2,4 battements par minute (bpm) par rapport à la valeur initiale (74 bpm) a été observée lors de la période contrôlée des études cliniques chez les adultes portant sur Bydureon BCise. Quinze pour cent des patients traités par l’exénatide à libération prolongée ont présenté des augmentations moyennes de la FC ≥ 10 bpm ; environ 5 % à 10 % des patients au sein des autres groupes de traitement ont présenté des augmentations moyennes de la FC ≥ 10 bpm.
Population pédiatrique
Le profil de sécurité de l'exénatide dans une étude clinique menée chez des adolescents et des enfants âgés de 10 ans ou plus (voir rubrique 5.1) était similaire à celui observé dans les études menées chez les adultes.
Dans l'étude pédiatrique, aucun événement majeur d'hypoglycémie n'a été observé.
Au cours de la période de traitement en double aveugle de 24 semaines, un patient (1,7 %) du groupe exénatide à libération prolongée et un patient (4,3 %) du groupe placebo ont présenté une hypoglycémie mineure (définie comme un épisode d'hypoglycémie non majeure présentant des symptômes compatibles avec une hypoglycémie et une valeur de glucose inférieure à 3 mmol/L [54 mg/dL] avant le traitement de l'épisode). Les deux patients recevaient de l'insuline comme traitement de fond.
D'autres événements d'hypoglycémie, des épisodes qui ne répondaient pas aux critères majeurs ou mineurs, ont été rapportés par l'investigateur chez 8 patients (13,6 %) et 1 patient (4,3 %) dans les groupes exénatide à libération prolongée et placebo, respectivement. Parmi ceux-ci, 6 patients du groupe exénatide à libération prolongée et 1 patient du groupe placebo ont reçu de l'insuline en traitement de fond.
Dans l'étude pédiatrique, le titre maximal d'anticorps obtenu à tout moment de l'étude était faible (<625) pour environ 29,3 % des patients et élevé (≥625) pour environ 63,8 % des patients. Le pourcentage de patients présentant des titres d'anticorps positifs a atteint un pic à la semaine 12 environ. Au fur et à mesure que l'étude se poursuivait jusqu'à la semaine 52, le pourcentage de patients présentant des titres élevés avait diminué (30,4 %) et le pourcentage de patients présentant des titres faibles (41,3 %) avait augmenté. Les patients présentant des titres d'anticorps élevés peuvent présenter une réponse atténuée de l'HbA1c (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance :
Site internet : www.notifieruneffetindesirable.be
e-mail : adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
AstraZeneca AB
SE-151 85 Södertälje
Suède
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/11/696/005-006
10. DATE DE MISE À JOUR DU TEXTE
11/2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3907169 | BYDUREON 2MG BCISE SUSP INJ LIB.PR.STYLO PREREM. 4 | A10BJ01 | € 91,54 | - | Oui | - | - |