1. DÉNOMINATION DU MÉDICAMENT
AMGEVITA 20 mg solution injectable en seringue préremplie
AMGEVITA 40 mg solution injectable en seringue préremplie
AMGEVITA 80 mg solution injectable en seringue préremplie
AMGEVITA 40 mg solution injectable en stylo prérempli
AMGEVITA 80 mg solution injectable en stylo prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
AMGEVITA 20 mg solution injectable en seringue préremplie
Une seringue unidose préremplie contient 20 mg d'adalimumab dans 0,2 mL de solution (100 mg/mL).
AMGEVITA 40 mg solution injectable en seringue préremplie
Une seringue unidose préremplie contient 40 mg d'adalimumab dans 0,4 mL de solution (100 mg/mL).
AMGEVITA 80 mg solution injectable en seringue préremplie
Une seringue unidose préremplie contient 80 mg d'adalimumab dans 0,8 mL de solution (100 mg/mL).
AMGEVITA 40 mg solution injectable en stylo prérempli
Un stylo unidose prérempli contient 40 mg d'adalimumab dans 0,4 mL de solution (100 mg/mL).
AMGEVITA 80 mg solution injectable en stylo prérempli
Un stylo unidose prérempli contient 80 mg d'adalimumab dans 0,8 mL de solution (100 mg/mL).
L'adalimumab est un anticorps monoclonal humain recombinant produit dans des cellules ovariennes de hamster chinois.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable (injection)
Solution injectable (injection) en stylo prérempli (SureClick)
Solution limpide et incolore à légèrement jaune.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Polyarthrite rhumatoïde
AMGEVITA en association au méthotrexate est indiqué pour :
- le traitement de la polyarthrite rhumatoïde modérément à sévèrement active de l'adulte lorsque la réponse aux traitements de fond (DMARD), y compris le méthotrexate, est inadéquate.
- le traitement de la polyarthrite rhumatoïde sévère, active et évolutive chez les adultes non précédemment traités par le méthotrexate.
AMGEVITA peut être donné en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement avec le méthotrexate est inadaptée.
AMGEVITA ralentit la progression des dommages structuraux articulaires mesurés par radiographie et améliore les capacités fonctionnelles lorsqu'il est administré en association au méthotrexate.
Arthrite juvénile idiopathique
Arthrite juvénile idiopathique polyarticulaire
AMGEVITA en association au méthotrexate est indiqué pour le traitement de l’arthrite juvénile idiopathique polyarticulaire évolutive chez les patients à partir de 2 ans en cas de réponse insuffisante à un ou plusieurs DMARD. AMGEVITA peut être administré en monothérapie en cas d’intolérance au méthotrexate ou lorsque la poursuite du traitement par le méthotrexate est inadaptée (pour l’efficacité en monothérapie, voir rubrique 5.1). L’adalimumab n’a pas été étudié chez les patients de moins de 2 ans.
Arthrite liée à l’enthésite
AMGEVITA est indiqué pour le traitement de l’arthrite active liée à l’enthésite chez les patients à partir de 6 ans en cas de réponse insuffisante ou d’intolérance au traitement conventionnel (voir rubrique 5.1).
Spondyloarthrite axiale
Spondylarthrite ankylosante (SA)
AMGEVITA est indiqué pour le traitement de la spondylarthrite ankylosante sévère et active chez l'adulte ayant eu une réponse inadéquate au traitement conventionnel.
Spondyloarthrite axiale sans signes radiographiques de SA
AMGEVITA est indiqué dans le traitement de la spondyloarthrite axiale sévère sans signes radiographiques de SA, mais avec des signes objectifs d’inflammation à l’IRM et/ou un taux élevé de CRP chez les adultes ayant eu une réponse inadéquate ou une intolérance aux anti-inflammatoires non stéroïdiens.
Rhumatisme psoriasique
AMGEVITA est indiqué pour le traitement du rhumatisme psoriasique actif et évolutif chez l’adulte lorsque la réponse à un DMARD antérieur a été inadéquate. AMGEVITA ralentit la progression des dommages structuraux articulaires périphériques tels que mesurés par radiographie, chez les patients ayant des formes polyarticulaires symétriques de la maladie (voir rubrique 5.1) et améliore les capacités fonctionnelles.
Psoriasis
AMGEVITA est indiqué dans le traitement du psoriasis en plaques, modéré à sévère, chez les patients adultes qui nécessitent un traitement systémique.
Psoriasis en plaques de l’enfant et l’adolescent
AMGEVITA est indiqué dans le traitement du psoriasis en plaques chronique sévère chez les enfants à partir de 4 ans et les adolescents en cas de réponse insuffisante à un traitement topique et aux photothérapies ou lorsque ces traitements sont inappropriés.
Hidrosadénite suppurée (HS)
AMGEVITA est indiqué dans le traitement de l'hidrosadénite suppurée (maladie de Verneuil) active, modérée à sévère, chez les adultes et les adolescents à partir de 12 ans en cas de réponse insuffisante au traitement systémique conventionnel de l’HS (voir rubriques 5.1 et 5.2).
Maladie de Crohn
AMGEVITA est indiqué dans le traitement de la maladie de Crohn active modérée à sévère, chez les patients adultes qui n’ont pas répondu malgré un traitement approprié et bien conduit par un corticoïde et/ou un immunosuppresseur ; ou chez lesquels ces traitements sont contre-indiqués ou mal tolérés.
Maladie de Crohn chez l'enfant et l’adolescent
AMGEVITA est indiqué dans le traitement de la maladie de Crohn active modérée à sévère, chez les enfants et les adolescents à partir de 6 ans, qui n'ont pas répondu à un traitement conventionnel comprenant un traitement nutritionnel de première intention et un corticoïde et/ou un immunomodulateur, ou chez lesquels ces traitements sont mal tolérés ou contre-indiqués.
Rectocolite hémorragique
AMGEVITA est indiqué dans le traitement de la rectocolite hémorragique active, modérée à sévère chez les patients adultes ayant eu une réponse inadéquate au traitement conventionnel, comprenant les corticoïdes et la 6‑mercaptopurine (6-MP) ou l'azathioprine (AZA), ou chez lesquels ces traitements sont contre-indiqués ou mal tolérés.
Rectocolite hémorragique chez l’enfant et l’adolescent
AMGEVITA est indiqué dans le traitement de la rectocolite hémorragique active, modérée à sévère chez les enfants et les adolescents à partir de 6 ans ayant eu une réponse inadéquate au traitement conventionnel, comprenant les corticoïdes et/ou la 6-mercaptopurine (6-MP) ou l’azathioprine (AZA), ou chez lesquels ces traitements sont mal tolérés ou contre-indiqués.
Uvéite
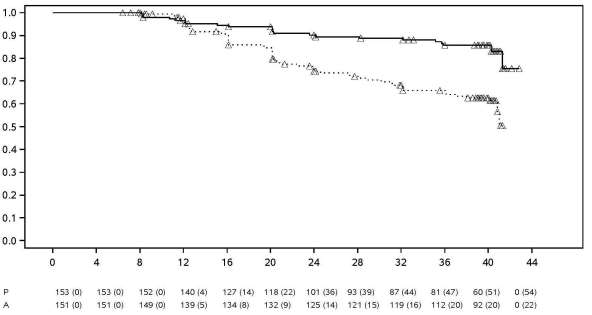
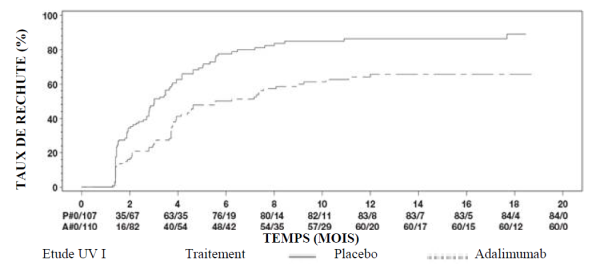
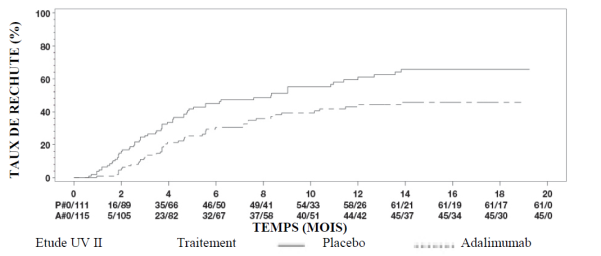
AMGEVITA est indiqué dans le traitement de l’uvéite non-infectieuse, intermédiaire, postérieure et de la panuvéite chez les patients adultes ayant eu une réponse insuffisante à la corticothérapie, chez les patients nécessitant une épargne cortisonique, ou chez lesquels la corticothérapie est inappropriée.
Uvéite chez l’enfant et l’adolescent
AMGEVITA est indiqué dans le traitement de l’uvéite antérieure chronique non infectieuse chez les enfants et les adolescents à partir de 2 ans en cas de réponse insuffisante ou d’intolérance au traitement conventionnel ou pour lesquels un traitement conventionnel est inapproprié.
4.2 Posologie et mode d'administration
Le traitement par AMGEVITA doit être instauré et supervisé par un médecin spécialiste qualifié en matière de diagnostic et de traitement des pathologies dans lesquelles AMGEVITA est indiqué. Il est recommandé aux ophtalmologistes de consulter un spécialiste approprié avant d’instaurer un traitement par AMGEVITA (voir rubrique 4.4). Une carte spéciale de surveillance sera remise aux patients traités par AMGEVITA.
Après une formation correcte à la technique d'injection, les patients peuvent s'auto-injecter AMGEVITA, si leur médecin l’estime possible, sous le couvert d’un suivi médical approprié.
Pendant le traitement par AMGEVITA, les autres traitements concomitants (tels que les corticoïdes et/ou immunomodulateurs) devront être optimisés.
Posologie
Polyarthrite rhumatoïde
Chez les patients adultes atteints de polyarthrite rhumatoïde, la posologie recommandée d'AMGEVITA est une dose unique de 40 mg d'adalimumab administrée toutes les deux semaines, par voie sous‑cutanée. L'administration de méthotrexate doit être continuée pendant le traitement par AMGEVITA.
Les glucocorticoïdes, les salicylés, les anti-inflammatoires non stéroïdiens ou les antalgiques peuvent être poursuivis pendant le traitement par AMGEVITA. En ce qui concerne l'association aux autres médicaments anti-rhumatismaux de fond autres que le méthotrexate voir rubriques 4.4 et 5.1.
En monothérapie, certains patients chez qui l'on observe une diminution de leur réponse à AMGEVITA 40 mg toutes les deux semaines, peuvent bénéficier d'une augmentation de la posologie à 40 mg d'adalimumab toutes les semaines ou 80 mg toutes les deux semaines.
Les données disponibles de l’adalimumab laissent supposer que la réponse clinique est habituellement obtenue en 12 semaines de traitement. La poursuite du traitement devra être reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Interruption du traitement
Il peut être nécessaire d’interrompre le traitement, par exemple avant une intervention chirurgicale ou en cas d’infection sévère.
Les données disponibles suggèrent que la ré-introduction de l’adalimumab après un arrêt de 70 jours ou plus a entraîné une réponse clinique de même ampleur et un profil de tolérance similaire à celui observé avant l’interruption du traitement.
Spondylarthrite ankylosante, spondyloarthrite axiale sans signes radiographiques de SA et rhumatisme psoriasique
La posologie recommandée d'AMGEVITA pour les patients atteints de spondylarthrite ankylosante, de spondyloarthrite axiale sans signes radiographiques de SA et pour les patients atteints de rhumatisme psoriasique est de 40 mg d'adalimumab en dose unique toutes les deux semaines, en injection sous‑cutanée.
Les données disponibles laissent supposer que la réponse clinique est habituellement obtenue en 12 semaines de traitement. La poursuite du traitement devra être reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Psoriasis
La posologie recommandée d’AMGEVITA pour débuter le traitement chez l’adulte est de 80 mg par voie sous‑cutanée. La posologie se poursuivra une semaine après par 40 mg en voie sous-cutanée une semaine sur deux.
La poursuite du traitement au-delà de 16 semaines doit être soigneusement reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Au-delà de 16 semaines, en cas de réponse insuffisante à AMGEVITA 40 mg toutes les deux semaines, les patients peuvent bénéficier d'une augmentation de la posologie à 40 mg toutes les semaines ou 80 mg toutes les deux semaines. Les bénéfices et les risques d’un traitement continu de 40 mg toutes les semaines ou 80 mg toutes les deux semaines doivent être soigneusement reconsidérés chez un patient en cas de réponse insuffisante après l’augmentation de la posologie (voir rubrique 5.1). En cas de réponse suffisante obtenue avec 40 mg toutes les semaines ou 80 mg toutes les deux semaines, la posologie peut ensuite être réduite à 40 mg toutes les deux semaines.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Hidrosadénite suppurée
Le schéma posologique recommandé d'AMGEVITA chez les patients adultes atteints d'hidrosadénite suppurée (HS) est d'une dose initiale de 160 mg au Jour 1 (administrée sous forme de 2 injections de 80 mg sur un jour ou de 1 injection de 80 mg par jour pendant deux jours consécutifs), suivie d'une dose de 80 mg deux semaines après au Jour 15. Deux semaines plus tard (Jour 29), poursuivre avec une dose de 40 mg toutes les semaines ou 80 mg toutes les deux semaines. Si nécessaire, les antibiotiques peuvent être poursuivis au cours du traitement par AMGEVITA. Au cours du traitement par AMGEVITA, il est recommandé au patient de nettoyer quotidiennement ses lésions avec un antiseptique topique.
La poursuite du traitement au-delà de 12 semaines doit être soigneusement reconsidérée chez les patients ne présentant pas d'amélioration pendant cette période.
Si le traitement est interrompu, AMGEVITA 40 mg toutes les semaines ou 80 mg toutes les deux semaines pourrait être réintroduit (voir rubrique 5.1).
Le bénéfice et le risque d'un traitement continu à long terme doivent faire l'objet d'une évaluation régulière (voir rubrique 5.1).
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Maladie de Crohn
Chez les patients adultes atteints de maladie de Crohn active modérée à sévère, le schéma posologique d'induction recommandé d'AMGEVITA est de 80 mg à la semaine 0, suivis de 40 mg à la semaine 2. S'il est nécessaire d'obtenir une réponse plus rapide au traitement, le schéma 160 mg à la semaine 0 (administrés sous forme de 2 injections de 80 mg par jour ou de 1 injection de 80 mg par jour pendant deux jours consécutifs), puis 80 mg à la semaine 2, peut être utilisé sachant que le risque d'événements indésirables est alors plus élevé pendant cette phase d'induction.
Après le traitement d'induction, la posologie recommandée est une dose de 40 mg administrée toutes les deux semaines, en injection sous-cutanée. Si un patient a arrêté le traitement par AMGEVITA et si les signes et symptômes de la maladie réapparaissent, AMGEVITA pourra être ré-administré. L'expérience de la ré‑administration du traitement au-delà de 8 semaines après la dose précédente est limitée.
Pendant le traitement d'entretien, les corticoïdes pourront être progressivement diminués conformément aux recommandations de pratique clinique.
Certains patients chez qui une diminution de la réponse au traitement par AMGEVITA 40 mg toutes les deux semaines est observée peuvent bénéficier d'une augmentation de la posologie à 40 mg d'AMGEVITA toutes les semaines ou 80 mg toutes les deux semaines.
Certains patients n’ayant pas répondu au traitement à la semaine 4 pourront poursuivre le traitement d’entretien jusqu’à la semaine 12. La poursuite du traitement devra être soigneusement reconsidérée chez un patient n’ayant pas répondu dans ces délais.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Rectocolite hémorragique
Chez les patients adultes atteints de rectocolite hémorragique modérée à sévère, le schéma posologique d'induction recommandé d'AMGEVITA est de 160 mg à la semaine 0 (administrés sous forme de 2 injections de 80 mg par jour ou de 1 injection de 80 mg par jour pendant deux jours consécutifs) et de 80 mg à la semaine 2. Après le traitement d'induction, la posologie recommandée est de 40 mg toutes les deux semaines, en injection sous-cutanée.
Pendant le traitement d'entretien, les corticoïdes pourront être progressivement diminués conformément aux recommandations de pratique clinique.
Certains patients chez qui une diminution de la réponse au traitement par AMGEVITA 40 mg toutes les deux semaines est observée pourront bénéficier d'une augmentation de la posologie à 40 mg d'AMGEVITA toutes les semaines ou 80 mg toutes les deux semaines.
Les données disponibles suggèrent que la réponse clinique est habituellement obtenue en 2 à 8 semaines de traitement. Le traitement par AMGEVITA ne doit pas être poursuivi chez les patients n'ayant pas répondu dans ces délais.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Uvéite
Chez les patients adultes atteints d’uvéite, la posologie recommandée d’AMGEVITA est d’une dose initiale de 80 mg suivie d’une dose de 40 mg toutes les deux semaines en commençant une semaine après l’administration de la première dose. L’expérience sur l’instauration du traitement par l’adalimumab en monothérapie est limitée. Le traitement par AMGEVITA peut être débuté en association avec une corticothérapie et/ou avec d’autres traitements immunomodulateurs non biologiques. La dose de corticoïdes associée peut être progressivement diminuée conformément à la pratique clinique, en débutant deux semaines après l’instauration du traitement par AMGEVITA.
Une réévaluation annuelle des bénéfices et des risques associés au traitement continu à long terme est recommandée (voir rubrique 5.1).
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Populations particulières
Sujets âgés
Aucun ajustement de la posologie n'est nécessaire.
Insuffisants rénaux et/ou hépatiques
L’adalimumab n'a pas été étudié dans ces populations de patients. Il n'est pas possible de recommander des posologies.
Population pédiatrique
Arthrite juvénile idiopathique
Arthrite juvénile idiopathique polyarticulaire à partir de 2 ans
La posologie recommandée d’AMGEVITA pour les patients atteints d’arthrite juvénile idiopathique polyarticulaire à partir de l’âge de 2 ans dépend du poids corporel (tableau 1). AMGEVITA est administré toutes les deux semaines en injection sous-cutanée.
Tableau 1. Posologie d’AMGEVITA chez les patients atteints d’arthrite juvénile idiopathique polyarticulaire
Poids du patient | Schéma posologique |
10 kg à < 30 kg | 20 mg toutes les deux semaines |
≥ 30 kg | 40 mg toutes les deux semaines |
Les données cliniques disponibles suggèrent que la réponse clinique est habituellement obtenue en 12 semaines de traitement. La poursuite du traitement devra être soigneusement reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Il n’y a pas d’utilisation justifiée d’adalimumab chez les patients âgés de moins de 2 ans dans cette indication.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Arthrite liée à l’enthésite
La posologie recommandée d’AMGEVITA pour les patients atteints d’arthrite liée à l’enthésite à partir de l’âge de 6 ans dépend du poids corporel (tableau 2). AMGEVITA est administré toutes les deux semaines en injection sous-cutanée.
Tableau 2. Posologie d’AMGEVITA chez les patients atteints d’arthrite liée à l’enthésite
Poids du patient | Schéma posologique |
15 kg à < 30 kg | 20 mg toutes les deux semaines |
≥ 30 kg | 40 mg toutes les deux semaines |
L’adalimumab n’a pas été étudié chez les patients de moins de 6 ans atteints d’arthrite liée à l’enthésite.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Rhumatisme psoriasique et spondyloarthrite axiale y compris spondylarthrite ankylosante
Il n’y a pas d’utilisation justifiée d’adalimumab dans la population pédiatrique dans les indications, spondylarthrite ankylosante et rhumatisme psoriasique.
Psoriasis en plaques de l’enfant et l’adolescent
La posologie recommandée d’AMGEVITA pour les patients atteints de psoriasis en plaques âgés de 4 à 17 ans dépend du poids corporel (tableau 3). AMGEVITA est administré en injection sous-cutanée.
Tableau 3. Posologie d’AMGEVITA chez les enfants et les adolescents atteints de psoriasis en plaques
Poids du patient | Schéma posologique |
15 kg à < 30 kg | Dose initiale de 20 mg puis 20 mg toutes les deux semaines en commençant une semaine après l’administration de la dose initiale |
≥ 30 kg | Dose initiale de 40 mg puis 40 mg toutes les deux semaines en commençant une semaine après l’administration de la dose initiale |
La poursuite du traitement au-delà de 16 semaines doit être soigneusement reconsidérée chez un patient n’ayant pas répondu dans ces délais.
Si un retraitement par AMGEVITA est indiqué, les recommandations ci-dessus pour la posologie et la durée de traitement doivent être suivies.
La sécurité d’adalimumab chez l’enfant et l’adolescent présentant un psoriasis en plaques a été évaluée sur une durée moyenne de 13 mois.
Il n’y a pas d’utilisation justifiée d’AMGEVITA chez les enfants âgés de moins de 4 ans dans cette indication.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Hidrosadénite suppurée de l’adolescent (à partir de 12 ans, pesant au moins 30 kg)
Il n’existe pas d’essai clinique conduit avec adalimumab chez des adolescents atteints d’HS. La posologie d’AMGEVITA chez ces patients a été déterminée à partir d’une modélisation pharmacocinétique et d’une simulation (voir rubrique 5.2).
La posologie recommandée d’AMGEVITA est de 80 mg à la semaine 0 suivie de 40 mg toutes les deux semaines à partir de la semaine 1 en injection sous-cutanée.
Chez les adolescents avec une réponse insuffisante à AMGEVITA 40 mg toutes les deux semaines, une augmentation de la posologie à 40 mg toutes les semaines ou 80 mg toutes les deux semaines peut être envisagée.
Si nécessaire, les antibiotiques peuvent être poursuivis au cours du traitement par AMGEVITA. Au cours du traitement par AMGEVITA, il est recommandé au patient de nettoyer quotidiennement ses lésions avec un antiseptique topique.
La poursuite du traitement au-delà de 12 semaines doit être soigneusement reconsidérée chez les patients ne présentant pas d’amélioration pendant cette période.
Si le traitement est interrompu, AMGEVITA pourrait être réintroduit si nécessaire.
Le bénéfice et le risque d’un traitement continu à long terme doivent faire l’objet d’une évaluation régulière (voir les données chez les adultes à la rubrique 5.1).
Il n’y a pas d’utilisation justifiée d’AMGEVITA chez les enfants âgés de moins de 12 ans dans cette indication.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Maladie de Crohn de l'enfant et l'adolescent
La posologie recommandée d'AMGEVITA pour les patients atteints de la maladie de Crohn âgés de 6 à 17 ans dépend du poids corporel (tableau 4). AMGEVITA est administré en injection sous-cutanée.
Tableau 4. Posologie d’AMGEVITA chez les enfants et les adolescents atteints de la maladie de Crohn
Poids du patient | Dose d’induction | Dose d’entretien à partir de la semaine 4 |
< 40 kg |
| 20 mg toutes les deux semaines |
≥ 40 kg |
| 40 mg toutes les deux semaines |
Les patients chez qui une réponse insuffisante au traitement est observée peuvent bénéficier d'une augmentation de la posologie :
- < 40 kg : 20 mg toutes les semaines
- ≥ 40 kg : 40 mg toutes les semaines ou 80 mg toutes les deux semaines
La poursuite du traitement devra être soigneusement reconsidérée chez un patient n'ayant pas répondu à la semaine 12.
Il n’y a pas d’utilisation justifiée d’adalimumab chez les enfants âgés de moins de 6 ans dans cette indication.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Rectocolite hémorragique chez l’enfant et l’adolescent
La posologie recommandée d’AMGEVITA pour les patients âgés de 6 à 17 ans et atteints de rectocolite hémorragique dépend du poids corporel (tableau 5). AMGEVITA est administré par injection sous‑cutanée.
Tableau 5. Posologie d’AMGEVITA chez les enfants et les adolescents atteints de rectocolite hémorragique
Poids du patient | Dose d’induction | Dose d’entretien à partir de la semaine 4* |
< 40 kg |
| 40 mg toutes les deux semaines |
≥ 40 kg |
| 80 mg toutes les deux semaines |
* Pour les patients atteignant l’âge de 18 ans pendant le traitement par AMGEVITA, la dose d’entretien prescrite doit être maintenue.
La poursuite du traitement au‑delà de 8 semaines doit être soigneusement reconsidérée chez les patients n’ayant pas répondu pendant cette période.
Il n’y a pas d’utilisation justifiée d’AMGEVITA chez les enfants âgés de moins de 6 ans dans cette indication.
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Uvéite chez l’enfant et l’adolescent
La posologie recommandée d’AMGEVITA pour les enfants et les adolescents atteints d’uvéite à partir de l’âge de 2 ans dépend du poids corporel (tableau 6). AMGEVITA est administré en injection sous-cutanée.
Dans l’uvéite chez l’enfant et l’adolescent, aucun essai clinique n’a été conduit avec AMGEVITA sans traitement concomitant par le méthotrexate.
Tableau 6. Posologie d’AMGEVITA chez les enfants et les adolescents atteints d’uvéite
Poids du patient | Schéma posologique |
< 30 kg | 20 mg toutes les deux semaines en association avec du méthotrexate |
≥ 30 kg | 40 mg toutes les deux semaines en association avec du méthotrexate |
Lors de l’instauration du traitement par AMGEVITA, une dose de charge de 40 mg pour les patients < 30 kg ou de 80 mg pour les patients ≥ 30 kg peut être administrée une semaine avant le début du traitement d’entretien. Aucune donnée clinique n’est disponible sur l’utilisation d’une dose de charge d’AMGEVITA chez les enfants âgés de moins de 6 ans (voir rubrique 5.2).
Il n’y a pas d’utilisation justifiée d’AMGEVITA chez les enfants âgés de moins de 2 ans dans cette indication.
Une réévaluation annuelle des bénéfices et des risques associés au traitement continu à long terme est recommandée (voir rubrique 5.1).
Différents dosages et/ou présentations d’AMGEVITA peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Mode d’administration
AMGEVITA est administré en injection sous-cutanée. Les instructions complètes d’utilisation sont fournies dans la notice.
4.3 Contre-indications
Hypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique 6.1.
Tuberculose évolutive ou autres infections sévères telles que sepsis et infections opportunistes (voir rubrique 4.4).
Insuffisance cardiaque modérée à sévère (NYHA classes III/IV) (voir rubrique 4.4).
4.8 Effets indésirables
Résumé du profil de tolérance
L’adalimumab a été étudié chez 9 506 patients dans des essais pivots contrôlés et en ouvert d’une durée de 60 mois et plus. Ces essais ont inclus des patients atteints de polyarthrite rhumatoïde récente ou ancienne, d’arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite liée à l’enthésite) ou des patients souffrant de spondyloarthrite axiale (spondylarthrite ankylosante et spondyloarthrite axiale sans signes radiographiques de SA), de rhumatisme psoriasique, de la maladie de Crohn, de rectocolite hémorragique, de psoriasis et d'hidrosadénite suppurée et d’uvéite. Les études contrôlées pivots portaient sur 6 089 patients ayant reçu de l’adalimumab et 3 801 patients ayant reçu un placebo ou un comparateur actif pendant la phase contrôlée.
Le pourcentage de patients ayant interrompu le traitement en raison d'effets indésirables pendant la phase en double aveugle, contrôlée, des études pivots a été de 5,9 % chez les patients traités par l’adalimumab et de 5,4 % chez les patients du groupe contrôle.
Les effets indésirables les plus fréquemment rapportés sont les infections (telles que les rhinopharyngites, les infections des voies respiratoires hautes et les sinusites), les réactions au site d’injection (érythème, démangeaisons, hémorragie, douleur ou gonflement), les céphalées et les douleurs musculo-squelettiques.
Des effets indésirables graves ont été rapportés avec l’adalimumab. Les antagonistes du TNF, tels qu’AMGEVITA affectent le système immunitaire et leur utilisation peut avoir des répercussions sur les défenses du corps contre les infections et le cancer.
Des infections menaçant le pronostic vital et d’issue fatale (comprenant sepsis, infections opportunistes et tuberculose), des réactivations d’hépatite B et différents cancers (y compris leucémie, lymphome et lymphome hépatosplénique à lymphocytes T) ont également été rapportés avec l’utilisation de l’adalimumab.
Des effets hématologiques, neurologiques et autoimmuns sévères ont été aussi rapportés. Ceci comprend de rares cas de pancytopénie, d’anémie médullaire, des cas de démyélinisation centrale et périphérique et des cas de lupus, d’événements liés au lupus et de syndrome de Stevens-Johnson.
Population pédiatrique
En général, la fréquence et le type des événements indésirables observés chez l’enfant et l’adolescent ont été comparables à ceux observés chez les patients adultes.
Liste des effets indésirables
La liste des effets indésirables est basée sur les études cliniques et sur l’expérience après commercialisation et est présentée par système-organe et par fréquence dans le tableau 7 ci-dessous : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) et indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. La fréquence la plus élevée observée dans les diverses indications a été incluse. La présence d’un astérisque (*) dans la colonne « Classe de systèmes d’organes » indique que de plus amples informations sont disponibles aux rubriques 4.3, 4.4 et 4.8.
Tableau 7. Effets indésirables
Classe de systèmes d’organes | Fréquence | Effets indésirables |
Infections et infestations* | Très fréquent | Infections des voies respiratoires (y compris infection des voies respiratoires basses et infection des voies respiratoires hautes, pneumonie, sinusite, pharyngite, rhino-pharyngite et pneumonie herpétique) |
| Fréquent | Infections systémiques (y compris sepsis, candidose et grippe), |
Peu fréquent | Infections neurologiques (y compris méningite virale), | |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes)* | Fréquent | Cancer de la peau à l’exclusion du mélanome (y compris carcinome basocellulaire et carcinome malpighien spino-cellulaire), |
Peu fréquent | Lymphome**, | |
Rare | Leucémie1) | |
Indéterminé | Lymphome hépatosplénique à lymphocytes T1), Carcinome à cellules de Merkel (carcinome neuroendocrine cutané)1), | |
Affections hématologiques et du système lymphatique* | Très fréquent | Leucopénie (y compris neutropénie et agranulocytose), |
Fréquent | Leucocytose, | |
Peu fréquent | Purpura thrombopénique idiopathique | |
Rare | Pancytopénie | |
Affections du système immunitaire* | Fréquent | Hypersensibilité, |
Peu fréquent | Sarcoïdose1), Vascularite | |
Rare | Anaphylaxie1) | |
Troubles du métabolisme et de la nutrition | Très fréquent | Augmentation du taux de lipides |
Fréquent | Hypokaliémie, | |
Affections psychiatriques | Fréquent | Troubles de l’humeur (y compris dépression), |
Affections du système nerveux* | Très fréquent | Céphalées |
Fréquent | Paresthésie (y compris hypoesthésie), | |
Peu fréquent | Accident vasculaire cérébral1), | |
Rare | Sclérose en plaques, | |
Affections oculaires | Fréquent | Troubles visuels, |
Peu fréquent | Diplopie | |
Affections de l'oreille et du labyrinthe | Fréquent | Vertiges |
Peu fréquent | Surdité, | |
Affections cardiaques* | Fréquent | Tachycardie |
Peu fréquent | Infarctus du myocarde1), | |
Rare | Arrêt cardiaque | |
Affections vasculaires | Fréquent | Hypertension, |
Peu fréquent | Anévrisme aortique, | |
Affections respiratoires, thoraciques et médiastinales* | Fréquent | Asthme, Dyspnée, Toux |
Peu fréquent | Embolie pulmonaire1), | |
Rare | Fibrose pulmonaire1) | |
Affections gastro- intestinales | Très fréquent | Douleurs abdominales, |
Fréquent | Hémorragie gastro-intestinale, Dyspepsie, | |
Peu fréquent | Pancréatite, | |
Rare | Perforation intestinale1) | |
Affections hépatobiliaires* | Très fréquent | Elévation des enzymes hépatiques |
Peu fréquent | Cholécystite et lithiase biliaire, | |
Rare | Hépatite, | |
Indéterminé | Insuffisance hépatique1) | |
Affections de la peau et du tissu sous-cutané | Très fréquent | Rash (y compris éruption exfoliative) |
Fréquent | Aggravation ou apparition d’un psoriasis (y compris psoriasis pustulaire palmoplantaire) 1), | |
Peu fréquent | Sueurs nocturnes, | |
Rare | Erythème polymorphe1), | |
Indéterminée | Aggravation des symptômes de dermatomyosite1) | |
Affections musculo- squelettiques et systémiques | Très fréquent | Douleurs musculo-squelettiques |
Fréquent | Spasmes musculaires (y compris augmentation de la créatine phosphokinase sérique) | |
Peu fréquent | Rhabdomyolyse, | |
Rare | Syndrome type lupus1) | |
Affections du rein et des voies urinaires | Fréquent | Insuffisance rénale, |
Peu fréquent | Nycturie | |
Affections des organes de reproduction et du sein | Peu fréquent | Troubles de la fonction érectile |
Troubles généraux et anomalies au site d'administration* | Très fréquent | Réaction au site d’injection (y compris érythème au site d’injection) |
| Fréquent | Douleur thoracique, |
Peu fréquent | Inflammation | |
Investigations* | Fréquent | Troubles de la coagulation et troubles hémorragiques (incluant un allongement du temps de céphaline activé), |
Indéterminée | Augmentation du poids2) | |
Lésions, intoxications et complications liées aux procédures | Fréquent | Mauvaise cicatrisation |
* de plus amples informations sont disponibles aux rubriques 4.3, 4.4 et 4.8.
** y compris les études d’extension en ouvert.
1) Comprenant les données des notifications spontanées.
2) Le changement de poids moyen par rapport aux valeurs initiales pour l’adalimumab allait de 0,3 kg à 1,0 kg pour toutes les indications chez l’adulte, contre (moins) -0,4 kg à 0,4 kg pour le placebo, sur une période de traitement de 4 à 6 mois. Une augmentation de poids comprise entre 5 et 6 kg a également été observée au cours d’études d’extension à long terme, avec des expositions moyennes d’environ 1 à 2 ans sans groupe témoin, en particulier chez les patients atteints de la maladie de Crohn et de colite ulcéreuse. Le mécanisme qui sous-tend cet effet n’est pas clair mais pourrait être associé à l’action anti-inflammatoire de l’adalimumab.
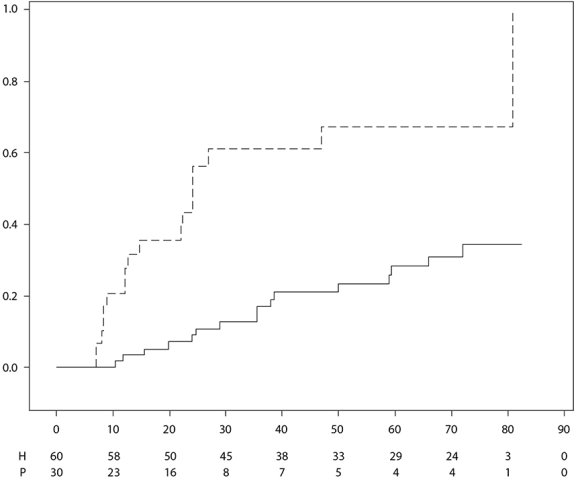
Hidrosadénite suppurée (HS)
Le profil de sécurité chez les patients atteints d'HS traités par l’adalimumab de façon hebdomadaire correspond au profil de sécurité connu de l’adalimumab.
Uvéite
Le profil de sécurité chez les patients atteints d’uvéite traités par l’adalimumab toutes les deux semaines correspond au profil de sécurité connu de l’adalimumab.
Description des effets indésirables sélectionnés
Réactions au point d'injection
Dans les essais contrôlés pivots menés chez l'adulte et l'enfant, 12,9 % des patients traités par l’adalimumab ont présenté des réactions au point d'injection (érythème et/ou prurit, saignement, douleur ou tuméfaction) contre 7,2 % des patients recevant le placebo ou le comparateur actif. Les réactions au point d'injection n'ont généralement pas nécessité l'arrêt du médicament.
Infections
Dans les essais contrôlés pivots menés chez l'adulte et l'enfant, la fréquence des infections a été de 1,51 par patient-année dans le groupe adalimumab et de 1,46 par patient-année dans le groupe placebo et le groupe contrôle. Les infections consistaient essentiellement en nasopharyngites, infections de l'appareil respiratoire supérieur et sinusites. La plupart des patients ont continué l’adalimumab après la guérison de l'infection.
L’incidence des infections graves a été de 0,04 par patient-année dans le groupe adalimumab et de 0,03 par patient-année dans le groupe placebo et le groupe contrôle.
Dans les études contrôlées et en ouvert avec l’adalimumab menées chez l'adulte et dans la population pédiatrique, des infections graves (y compris des infections à issue fatale, ce qui s'est produit rarement) ont été rapportées dont des signalements de tuberculose (y compris miliaire et à localisations extra-pulmonaires) et d'infections opportunistes invasives (par ex. histoplasmose disséminée ou histoplasmose extrapulmonaire, blastomycose, coccidioïdomycose, pneumocystose, candidose, aspergillose et listériose). La plupart des cas de tuberculose sont survenus dans les huit premiers mois après le début du traitement et peuvent être le reflet d'une réactivation d'une maladie latente.
Tumeurs malignes et troubles lymphoprolifératifs
Aucun cas de cancer n’a été observé chez 249 patients pédiatriques représentant une exposition de 655,6 patient-années lors des études de l’adalimumab chez les patients atteints d’arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite liée à l’enthésite). De plus, aucun cas de cancer n’a été observé chez 192 patients pédiatriques représentant une exposition de 498,1 patient-années lors des études avec l’adalimumab dans la maladie de Crohn pédiatrique. Aucun cas de cancer n’a été observé chez 77 patients pédiatriques correspondant à une exposition de 80 patient-années lors d’une étude avec l’adalimumab dans le psoriasis en plaques chronique pédiatrique. Lors d’une étude avec adalimumab menée chez des enfants et des adolescents atteints de rectocolite hémorragique, aucun cas de cancer n’a été observé chez 93 enfants et adolescents représentant une exposition de 65,3 patient‑années. Aucun cas de cancer n’a été observé chez 60 patients pédiatriques représentant une exposition de 58,4 patient-années lors d’une étude avec adalimumab dans l’uvéite pédiatrique.
Pendant les périodes contrôlées des essais cliniques pivots chez l'adulte avec l’adalimumab d’une durée d’au moins 12 semaines chez des patients souffrant de polyarthrite rhumatoïde modérément à sévèrement active, de spondylarthrite ankylosante, de spondyloarthrite axiale sans signes radiographiques de SA, de rhumatisme psoriasique, de psoriasis, d’hidrosadénite suppurée, de la maladie de Crohn, de rectocolite hémorragique et d’uvéite, un taux (intervalle de confiance 95 %) de cancers autres que lymphomes ou cancers de la peau non mélanomes, de 6,8 (4,4 – 10,5) pour 1 000 patient-années parmi les 5 291 patients traités par l’adalimumab, a été observé versus un taux de 6,3 (3,4 – 11,8) pour 1 000 patient-années parmi les 3 444 patients du groupe contrôle (la durée moyenne du traitement était de 4,0 mois pour les patients traités par l’adalimumab et de 3,8 mois pour les patients du groupe contrôle). Le taux (intervalle de confiance de 95 %) de cancers de la peau non‑mélanomes était de 8,8 (6,0 – 13,0) pour 1 000 patient-années pour les patients traités par l’adalimumab et de 3,2 (1,3 – 7,6) pour 1 000 patient-années parmi les patients du groupe contrôle. Dans ces cancers de la peau, les carcinomes spino-cellulaires sont survenus à des taux de 2,7 (1,4 – 5,4) pour 1 000 patient-années chez les patients traités par l’adalimumab et 0,6 (0,1 – 4,5) pour 1 000 patient-années chez les patients du groupe contrôle (intervalle de confiance 95 %). Le taux (intervalle de confiance 95 %) de lymphomes était de 0,7 (0,2 – 2,7) pour 1 000 patient-années chez les patients traités par l’adalimumab et 0,6 (0,1 ‑ 4,5) pour 1 000 patient-années chez les patients du groupe contrôle.
En joignant les périodes contrôlées de ces essais et les essais d'extension en ouvert d’adalimumab terminés ou en cours avec une durée moyenne d'environ 3,3 ans incluant 6 427 patients et plus de 26 439 patient-années de traitement, le taux observé de cancers, autres que lymphomes et cancers de la peau non mélanomes est d'environ 8,5 pour 1 000 patient-années. Le taux observé de cancers de la peau non‑mélanomes est d'environ 9,6 pour 1 000 patient-années et le taux de lymphomes observés est d'environ 1,3 pour 1 000 patient-années.
En post-marketing de janvier 2003 à décembre 2010, essentiellement chez les patients atteints de polyarthrite rhumatoïde, le taux spontanément rapporté de cancers est approximativement de 2,7 pour 1 000 patient‑années de traitement. Les taux spontanément rapportés pour les cancers de la peau non-mélanomes et les lymphomes sont respectivement d'environ 0,2 et 0,3 pour 1 000 patient-années de traitement (voir rubrique 4.4).
Au cours de la surveillance post-marketing, de rares cas de lymphome hépatosplénique à lymphocytes T ont été rapportés chez des patients traités par l’adalimumab (voir rubrique 4.4).
Auto-anticorps
Des recherches d'auto-anticorps répétées ont été effectuées sur des échantillons de sérum des patients des essais I-V dans la polyarthrite rhumatoïde. Dans ces essais, les titres d'anticorps antinucléaires initialement négatifs étaient positifs à la semaine 24 chez 11,9 % des patients traités par l’adalimumab et 8,1 % des patients sous placebo et comparateur. Deux patients sur les 3 441 traités par l’adalimumab dans toutes les études dans la polyarthrite rhumatoïde et le rhumatisme psoriasique ont présenté des signes cliniques évoquant un syndrome pseudo-lupique d'apparition nouvelle. L'état des patients s'est amélioré après l'arrêt du traitement. Aucun patient n'a présenté de néphrite lupique ou de symptômes nerveux centraux.
Evénements hépatobiliaires
Dans les essais cliniques contrôlés de phase III d’adalimumab dans la polyarthrite rhumatoïde et le rhumatisme psoriasique avec une période de contrôle de 4 à 104 semaines, des élévations d'ALAT ≥ 3 × N sont survenues chez 3,7% des patients traités par l’adalimumab et chez 1,6% des patients du groupe contrôle.
Dans les essais cliniques contrôlés de phase III d’adalimumab chez les patients atteints d’arthrite juvénile idiopathique polyarticulaire âgés de 4 à 17 ans et les patients atteints d’arthrite liée à l’enthésite âgés de 6 à 17 ans, des élévations d’ALAT ≥ 3 × N sont survenues chez 6,1 % des patients traités par l’adalimumab et chez 1,3 % des patients du groupe contrôle. La plupart des élévations d’ALAT sont survenues dans le cadre d’une utilisation concomitante de méthotrexate. Aucune élévation d’ALAT ≥ 3 × N n’est survenue au cours de l’essai de phase III d’adalimumab chez des patients atteints d’arthrite juvénile idiopathique polyarticulaire âgés de 2 à < 4 ans.
Dans les essais cliniques contrôlés de phase III d’adalimumab chez les patients atteints de maladie de Crohn et de rectocolite hémorragique avec une période de contrôle de 4 à 52 semaines, des élévations d'ALAT ≥ 3 × N sont survenues chez 0,9% des patients traités par l’adalimumab et chez 0,9% des patients du groupe contrôle.
Dans l’essai clinique de phase III d’adalimumab chez les enfants et adolescents atteints de maladie de Crohn qui a évalué l’efficacité et le profil de sécurité de deux schémas posologiques d’entretien en fonction du poids après un traitement d’induction ajusté au poids jusqu’à 52 semaines de traitement, des élévations d’ALAT ≥ 3 × N sont survenues chez 2,6 % des patients (5/192), parmi lesquels 4 étaient traités en association avec des immunosuppresseurs au début de l’étude.
Dans les essais cliniques contrôlés de phase III d’adalimumab dans le psoriasis en plaques avec une période de contrôle de 12 à 24 semaines, des élévations d'ALAT ≥ 3 × N sont survenues chez 1,8 % des patients traités par l’adalimumab et chez 1,8 % des patients du groupe contrôle.
Il n’a pas été observé d’élévations de l’ALAT ≥ 3 × N dans l’étude de phase III d’adalimumab chez des patients pédiatriques atteints de psoriasis en plaques.
Dans les essais cliniques contrôlés d'adalimumab (doses initiales de 160 mg à la Semaine 0 et 80 mg à la Semaine 2 suivies de 40 mg chaque semaine à partir de la semaine 4), chez les patients atteints d'hidrosadénite suppurée avec une période de contrôle de 12 à 16 semaines, des élévations d'ALAT ≥ 3 × N sont survenues chez 0,3 % des patients traités par l’adalimumab et 0,6 % des patients du groupe contrôle.
Dans les essais cliniques contrôlés d’adalimumab (dose initiale de 80 mg à la semaine 0 suivie de 40 mg toutes les deux semaines à partir de la semaine 1) chez les patients adultes atteints d’uvéite pour une durée allant jusqu’à 80 semaines, avec une durée médiane d’exposition de respectivement 166,5 jours et 105,0 jours pour les patients traités par l’adalimumab et les patients du groupe contrôle, des élévations d’ALAT ≥ 3 × N sont survenues chez 2,4 % des patients traités par l’adalimumab et 2,4 % des patients du groupe contrôle.
Dans l’essai clinique contrôlé de phase III d’adalimumab mené chez des enfants et des adolescents atteints de rectocolite hémorragique (N = 93) qui a évalué l’efficacité et le profil de sécurité d’une dose d’entretien de 0,6 mg/kg (dose maximale de 40 mg) administrée une semaine sur deux (N = 31) et d’une dose d’entretien de 0,6 mg/kg (dose maximale de 40 mg) administrée chaque semaine (N = 32), à la suite d’une dose d’induction ajustée en fonction du poids corporel de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 (N = 63), ou d’une dose d’induction de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0, d’un placebo à la semaine 1, et d’une dose de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 (N = 30), des élévations de l’ALAT ≥ 3 × LSN sont survenues chez 1,1 % (1/93) des patients.
Dans les essais cliniques, toutes indications confondues, les patients avec ALAT augmentées étaient asymptomatiques et dans la plupart des cas les élévations étaient transitoires et réversibles lors de la poursuite du traitement. Cependant, au cours de la surveillance post‑marketing, des insuffisances hépatiques ainsi que des désordres hépatiques moins sévères, qui peuvent précéder une insuffisance hépatique, tels que des hépatites y compris des hépatites auto-immunes, ont été rapportées chez des patients recevant de l’adalimumab.
Administration concomitante d’azathioprine/6-mercaptopurine
Lors d’études dans la maladie de Crohn chez l’adulte, une incidence plus élevée de tumeurs et d’infections graves a été observée avec l’association adalimumab et azathioprine/6-mercaptopurine comparativement à l’adalimumab utilisé seul.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance :
Site internet : www.notifieruneffetindesirable.be
e-mail : adr@afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHÉ
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
Pays-Bas
8. NUMÉROS D'AUTORISATION DE MISE SUR LE MARCHÉ
AMGEVITA 20 mg solution injectable en seringue préremplie
EU/1/16/1164/010 – 1 boîte
AMGEVITA 40 mg solution pour injection en seringue préremplie
EU/1/16/1164/011 – 1 boîte
EU/1/16/1164/012 – 2 boîtes
EU/1/16/1164/013 – 6 (3×2) conditionnement multiple
AMGEVITA 80 mg solution pour injection en seringue préremplie
EU/1/16/1164/017 – 1 boîte
EU/1/16/1164/018 – 2 boîtes
EU/1/16/1164/019 – 3 (3×1) conditionnement multiple
AMGEVITA 40 mg solution injectable en stylo prérempli
EU/1/16/1164/014 – 1 boîte
EU/1/16/1164/015 – 2 boîtes
EU/1/16/1164/016 – 6 (3×2) conditionnement multiple
AMGEVITA 80 mg solution injectable en stylo prérempli
EU/1/16/1164/020 – 1 boîte
EU/1/16/1164/021 – 2 boîtes
EU/1/16/1164/022 – 3 (3×1) conditionnement multiple
10. DATE DE MISE À JOUR DU TEXTE
octobre 2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4828471 | AMGEVITA 20MG SOL INJ 100MG/ML SER.PREREMPLIE 1 | € 121,55 | - | Oui | € 12,5 | € 8,3 | |
| 4828489 | AMGEVITA 40MG SOL INJ 100MG/ML SER.PREREMPLIE 2 | € 453,7 | - | Oui | € 12,5 | € 8,3 | |
| 4828497 | AMGEVITA 40MG SOL INJ 100MG/ML SER.PREREMPLIE 6 | € 1360,4 | - | Oui | € 12,5 | € 8,3 | |
| 4828505 | AMGEVITA 40MG SOL INJ 100MG/ML STYLO PREREMPLIE 2 | € 453,7 | - | Oui | € 12,5 | € 8,3 | |
| 4828513 | AMGEVITA 40MG SOL INJ 100MG/ML STYLO PREREMPLIE 6 | € 1360,4 | - | Oui | € 12,5 | € 8,3 | |
| 4828521 | AMGEVITA 80MG SOL INJ 100MG/ML STYLO PREREMPLIE 1 | € 453,7 | - | Oui | € 12,5 | € 8,3 |