RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
ADVATE 250 UI/5 mL poudre et solvant pour solution injectable
ADVATE 500 UI/5 mL poudre et solvant pour solution injectable
ADVATE 1 000 UI/5 mL poudre et solvant pour solution injectable
ADVATE 1 500 UI/5 mL poudre et solvant pour solution injectable
ADVATE 2 000 UI/5 mL poudre et solvant pour solution injectable
ADVATE 3 000 UI/5 mL poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
ADVATE 250 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 250 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 50 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 5 mL de solvant.
ADVATE 500 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 500 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 100 UI par mL de facteur VIII (ADNr) de coagulation humain, octocogalfa après reconstitution avec 5 mL de solvant.
ADVATE 1 000 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 1 000 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 200 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 5 mL de solvant.
ADVATE 1 500 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 1 500 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 300 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 5 mL de solvant.
ADVATE 2 000 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 2 000 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 400 UI par mL de facteur VIII ADNr) de coagulation humain, octocog alfa après reconstitution avec 5 mL de solvant.
ADVATE 3 000 UI/5 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 3 000 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 600 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 5 mL de solvant.
Le titre (Unité Internationale) est déterminé par dosage chromogénique, selon la Pharmacopée européenne. L’activité spécifique d’ADVATE est d'environ 4 520 ‑ 11 300 UI/mg de protéine.
L’octocog alfa (facteur VIII de coagulation humain (ADNr) est une protéine purifiée, qui a 2 332 acides aminés. Il est produit par la technique de l’ADN recombinant sur cellules d’ovaire dehamster chinois (CHO). Préparé sans addition de protéine (exogène) d’origine humaine ou animale lors des étapes de culture cellulaire, de purification ou de formulation finale.
Excipients à effet notoire
Ce médicament contient 0,45 mmol de sodium (10 mg) et 0,5 mg de polysorbate 80 par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Poudre et solvant pour solution injectable.
Poudre : friable de couleur blanche à légèrement grise.
Solvant : solution limpide et incolore.
Après reconstitution, la solution est limpide, incolore, exempte de particules étrangères et a un pH compris entre 6,7 et 7,3.
4. DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Traitement et prophylaxie des épisodes hémorragiques chez les patients atteints d’hémophilie A (déficit congénital en facteur VIII). ADVATE est indiqué dans tous les groupes d’âge.
4.2 Posologie et mode d’administration
Le traitement doit être instauré sous la surveillance d'un médecin expérimenté dans le traitement de l'hémophilie et avec une possibilité d'intervention immédiate en réanimation en cas d'anaphylaxie.
Surveillance thérapeutique
Au cours du traitement, il est conseillé de déterminer de façon appropriée les taux de facteur VIII afin d’évaluer la dose à administrer ainsi que la fréquence du renouvellement des injections. Selon les patients, la réponse au facteur VIII peut varier, entraînant des taux de récupération et des demi-vies différents. Il peut être nécessaire d’ajuster les doses basées sur le poids corporel chez les patients présentant une insuffisance pondérale ou un surpoids. Dans le cas d’interventions chirurgicales majeures en particulier, un contrôle précis du traitement substitutif par une analyse de l’activité coagulante (activité plasmatique du facteur VIII) est indispensable.
Posologie
La dose et la durée du traitement substitutif dépendent de la sévérité du déficit en facteur VIII, de la localisation et de l’importance de l’épisode hémorragique, ainsi que de l’état clinique du patient.
Le nombre d’unités de facteur VIII administrées est exprimé en Unités Internationales (UI), par rapport au standard actuel de l'OMS pour les concentrés de facteur VIII. L’activité coagulante en facteur VIII dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal), soit, de préférence, en Unités Internationales (par rapport au Standard International du facteur VIII plasmatique).
Une Unité Internationale (UI) d’activité du facteur VIII correspond à la quantité de facteur VIII contenue dans un mL de plasma humain normal.
Traitement à la demande
Le calcul de la dose nécessaire de facteur VIII est basé sur le résultat empirique qu'une UI de facteur VIII par kg de poids corporel augmente l'activité coagulante plasmatique du facteur VIII de 2 UI/dL. La dose nécessaire est déterminée à l'aide de la formule suivante :
Nombre d’unités (UI) nécessaires = poids corporel (kg) x augmentation souhaitée du taux de facteur VIII (%) x 0,5.
La dose et la fréquence d’administration doivent être adaptées à l’efficacité clinique chez chaque individu. Dans certaines circonstances (par exemple, présence d’un inhibiteur à faible titre), des doses plus importantes que les quantités calculées à l’aide de la formule peuvent être nécessaires.
En cas de survenue de l'un des événements hémorragiques suivants, l'activité du facteur VIII ne doit pas chuter en dessous du taux d'activité plasmatique indiqué (en % de la normale ou UI/dL) pendant la période mentionnée. Le tableau ci‑dessous (tableau 1) peut servir de guide pour la détermination des posologies lors d'épisodes hémorragiques et de chirurgie :
Tableau 1 : Guide pour la détermination de la posologie lors d’épisodes hémorragiques et de chirurgie | ||
Degré de l'hémorragie / type d'intervention chirurgicale | Niveau de facteur VIII nécessaire (% ou UI/dL) | Fréquence des doses (heures) / durée du traitement (jours) |
Hémorragie |
|
|
Début d’hémarthrose, de saignement musculaire ou buccal. | 20 – 40 | Renouveler les injections toutes les 12 à 24 heures (toutes les 8 à 24 heures chez les patients âgés de moins de 6 ans) pendant au moins 1 jour, jusqu'à la fin de l’épisode hémorragique, indiquée par la disparition de la douleur ou l'obtention d’une cicatrisation. |
Hémarthrose plus étendue, hémorragie musculaire ou hématome. | 30 – 60 | Renouveler les injections toutes les 12 à 24 heures (toutes les 8 à 24 heures chez les patients âgés de moins de 6 ans) pendant 3 – 4 jours ou plus jusqu’à disparition de la douleur et de l’invalidité aiguë. |
Hémorragie mettant en jeu le pronostic vital. | 60 – 100 | Répéter les injections toutes les 8 à 24 heures (toutes les 6 à 12 heures chez les patients âgés de moins de 6 ans) jusqu’à disparition du risque vital. |
Chirurgie |
|
|
Mineure | 30 – 60 | Toutes les 24 heures (toutes les 12 à 24 heures chez les patients âgés de moins de 6 ans), au moins 1 jour, jusqu’à l’obtention d'une cicatrisation. |
Majeure | 80 – 100 | Renouveler les injections toutes les 8 à 24 heures (toutes les 6 à 24 heures chez les patients âgés de moins de 6 ans) jusqu’à cicatrisation satisfaisante de la plaie, puis poursuivre le traitement pendant au moins 7 jours supplémentaires pour maintenir une activité coagulante du facteur VIII entre 30 % et 60 % (UI/dL). |
Prophylaxie
Pour le traitement prophylactique à long terme des épisodes hémorragiques chez les patients atteints d’hémophilie A sévère, les posologies habituelles sont de 20 à 40 UI de facteur VIII par kg de poids corporel à des intervalles de 2 à 3 jours.
Dans certains cas, surtout chez le sujet jeune, des intervalles plus rapprochés ou des doses plus élevées peuvent être nécessaires.
Population pédiatrique
La posologie pour le traitement à la demande des patients pédiatriques (entre 0 et 18 ans) ne diffère pas de celle des patients adultes. Chez les patients de moins de 6 ans, les doses recommandées pour un traitement prophylactique sont de 20 à 50 UI de facteur VIII par kg de poids corporel 3 à 4 fois par semaine.
Mode d’administration
Voie intraveineuse. En cas d’administration par un non-professionnel de santé, une formation appropriée est nécessaire.
La vitesse d'administration sera déterminée en fonction du niveau de confort du patient et jusqu'à un maximum de 10 mL/min.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
4.3 Contreindications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Réaction allergique connue aux protéines de hamster ou de souris.
4.8 Effets indésirables
Résumé du profil d'innocuité
Les études cliniques avec ADVATE comprenaient 418 sujets ayant connu au moins une exposition à ADVATE ; un total de 93 effets indésirables a été rapporté. Les effets indésirables qui ont été observés le plus fréquemment étaient le développement d’anticorps neutralisants du facteur VIII (inhibiteurs), des maux de tête et de la fièvre.
Une hypersensibilité ou des réactions allergiques (qui peuvent inclure : angioedème, brûlure et au site de perfusion, frissons, rougeurs, urticaire généralisée, céphalées, urticaire, hypotension, léthargie, nausées, impatience, tachycardie, oppression thoracique, foumillements, vomissements, sibilances) ont été rarement observées et peuvent, dans certains cas, évoluer vers une anaphylaxie sévère (y compris un choc).
L’apparition d’anticorps dirigés contre des protéines de souris et/ou de hamster peut être observée en rapport avec des réactions d’hypersensibilité.
Des anticorps neutralisants (inhibiteurs) peuvent apparaître chez des patients atteints d’hémophilie A traités avec le facteur VIII, y compris avec ADVATE (voir rubrique 5.1). Une telle apparition se manifeste par une réponse clinique insuffisante. Dans ce cas, il est recommandé de contacter un centre spécialisé en hémophilie.
Tableau de résumé des effets indésirables
Le tableau 2 présente la fréquence des effets indésirables issus des études cliniques et des signalements spontanés, conformément à la classification des systèmes d’organes MedDRA. (CSO et terme préconisé).
La fréquence a été évaluée selon les critères suivants : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/ 100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 : Fréquence des effets indésirables issus des études cliniques et des signalements spontanés | ||
Norme MedDRA | Effets indésirables | Fréquence des effets indésirablesa |
Infections et infestations | Grippe | Peu fréquent |
Laryngite | Peu fréquent | |
Affections hématologiques et du système lymphatique | Inhibition du facteur VIII | Peu fréquent (PPT)b |
Lymphangite | Peu fréquent | |
Affections du système immunitaire | Réaction anaphylactique* | Fréquence indéterminée |
Hypersensibilitéc* | Fréquence indéterminée | |
Affection du système nerveux | Maux de tête | Fréquent |
Vertiges | Peu fréquent | |
Troubles de la mémoire | Peu fréquent | |
Syncope | Peu fréquent | |
Tremblements | Peu fréquent | |
Migraine | Peu fréquent | |
Dysgueusie | Peu fréquent | |
Affections oculaires | Inflammation oculaire | Peu fréquent |
Affections cardiaques | Palpitations | Peu fréquent |
Affections vasculaires | Hématomes | Peu fréquent |
Bouffées de chaleur | Peu fréquent | |
Pâleur | Peu fréquent | |
Affections respiratoires, thoraciques et médiastinales | Dyspnées | Peu fréquent |
Affections gastro‑intestinales | Diarrhées | Peu fréquent |
Douleur abdominale haute | Peu fréquent | |
Nausées | Peu fréquent | |
Vomissements | Peu fréquent | |
Affections de la peau et du tissu sous‑cutané | Prurit | Peu fréquent |
Rash | Peu fréquent | |
Hyperhidrose | Peu fréquent | |
Urticaire | Peu fréquent | |
Troubles généraux et anomalies au site d’administration | Pyrexie | Fréquent |
Œdème périphérique | Peu fréquent | |
Douleur thoracique | Peu fréquent | |
Inconfort thoracique | Peu fréquent | |
Frissons | Peu fréquent | |
Etat anormal | Peu fréquent | |
Hématome au site de ponction vasculaire | Peu fréquent | |
Fatigue* | Fréquence indéterminée | |
Réaction au site d’injection* | Fréquence indéterminée | |
Malaise* | Fréquence indéterminée | |
Investigations | Augmentation du nombre de monocytes | Peu fréquent |
Diminution du facteur VIII de coagulationd | Peu fréquent | |
Diminution de l’hématocrite | Peu fréquent | |
Test biologique anormal | Peu fréquent | |
Lésions, intoxications et complications liées aux procédures | Complication post‑procédure | Peu fréquent |
Hémorragie post‑procédure | Peu fréquent | |
Réaction sur le site de l’intervention | Peu fréquent | |
a) Calculé sur la base du total de patients ayant reçu ADVATE (418) dans les études cliniques, à l’exception des effets indésirables identifiés dans le cadre de la surveillace post-commercialisation, marqués d’un *.
b) La fréquence est déterminée d’après des études portant sur tous les produits facteur VIII menées auprès de patients atteints d’hémophilie A sévère. PPT = patients précédemment traités, PUP = patients non traités précédemment (previously-untreated patients).
c) Effet indésirable expliqué dans la section ci-dessous.
d) La diminution inattendue de l’activité coagulante du facteur VIII est survenue chez un patient sous perfusion continue d’ADVATE après une chirurgie (jours post‑opératoires 10‑14). L’hémostase a été maintenue à tout moment pendant cette période. L’activité du facteur VIII et la clairance sont revenues à la normale à J15. Les recherches d’inhibiteur de facteur VIII réalisées à l’arrêt de la perfusion continue et à la fin de l’étude se sont révélées négatives.
Description de certains effets indésirables
Effets indésirables spécifiques aux résidus du processus de fabrication
Sur les 229 patients traités, pour lesquels le taux d’anticorps dirigés contre les protéines de cellules d’ovaire de hamster Chinois (CHO) a été évalué, ont été observés 3 cas d’augmentation statistiquement significative du titre, 4 cas de pics prolongés ou provisoires et un patient qui a présenté les deux à la fois mais sans aucun symptôme clinique. Sur les 229 patients traités, pour lesquels les anticorps dirigés contre les IgG d’origine murine ont été évalués, ont été observés 10 cas d’augmentation statistiquement significative des anticorps dirigés contre les antigènes murins, 2 cas de pics prolongés ou provisoires et un patient qui a présenté les deux à la fois. Chez quatre de ces patients, des cas isolés d’urticaire, de prurit, de rash et de numérations éosinophiles légèrement élevées ont été constatés au cours des expositions répétées au produit pendant l’étude.
Hypersensibilité
Les réactions de type allergique englobent l'anaphylaxie et se sont manifestées sous forme de vertiges, paresthésies, rash, bouffées congestives, gonflement facial, urticaire et prurit.
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables chez les enfants devraient être les mêmes que chez les adultes.
Outre le développement d'inhibiteurs chez des patients pédiatriques non préalablement traités (PUPs), et les complications liées au cathéter, aucune différence d'effets indésirables liée à l'âge n'a été constatée dans les études cliniques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Takeda Manufacturing Austria AG
Industriestrasse, 67
A‑1221 Vienne
Autriche
medinfoEMEA@takeda.com
8. NUMÉROS D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/03/271/001
EU/1/03/271/002
EU/1/03/271/003
EU/1/03/271/004
EU/1/03/271/005
EU/1/03/271/006
EU/1/03/271/011
EU/1/03/271/012
EU/1/03/271/013
EU/1/03/271/014
EU/1/03/271/015
EU/1/03/271/016
10. DATE DE MISE À JOUR DU TEXTE
05/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne du médicament https://www.ema.europa.eu.
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
ADVATE 250 UI/2 mL poudre et solvant pour solution injectable
ADVATE 500 UI/2 mL poudre et solvant pour solution injectable
ADVATE 1 000 UI/2 mL poudre et solvant pour solution injectable
ADVATE 1 500 UI/2 mL poudre et solvant pour solution injectable
ADVATE 250 UI/2 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 250 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 125 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 2 mL de solvant.
ADVATE 500 UI/2 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 500 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 250 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 2 mL de solvant.
ADVATE 1 000 UI/2 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 1 000 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 500 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 2 mL de solvant.
ADVATE 1 500 UI/2 mL poudre et solvant pour solution injectable
Chaque flacon contient nominalement 1 500 UI de facteur VIII (ADNr) de coagulation humain, octocog alfa. ADVATE contient approximativement 750 UI par mL de facteur VIII (ADNr) de coagulation humain, octocog alfa après reconstitution avec 2 mL de solvant.
Le titre (Unité Internationale) est déterminé par dosage chromogénique, selon la Pharmacopée européenne. L'activité spécifique d’ADVATE est d'environ 4 520 ‑ 11 300 UI/mg de protéine.
L’octocog alfa (facteur VIII de coagulation humain (ADNr)) est une protéine purifiée, qui a 2 332 acides aminés. Il est produit par la technique de l’ADN recombinant sur cellules d’ovaire de hamster chinois (CHO). Préparé sans addition de protéine (exogène) d’origine humaine ou animale lors des étapes de culture cellulaire, de purification ou de formulation finale.
Excipients à effet notoire
Ce médicament contient 0,45 mmol de sodium (10 mg) et 0,5 mg de polysorbate 80 par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable.
Poudre : friable de couleur blanche à légèrement grise.
Solvant : solution limpide et incolore.
Après reconstitution, la solution est limpide, incolore, exempte de particules étrangères et a un pH compris entre 6,7 et 7,3.
Traitement et prophylaxie des épisodes hémorragiques chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII). ADVATE est indiqué dans tous les groupes d'âge.
Le traitement doit être instauré sous la surveillance d'un médecin expérimenté dans le traitement de l'hémophilie et avec une possibilité d'intervention immédiate en réanimation en cas d'anaphylaxie.
Surveillance thérapeutique
Au cours du traitement, il est conseillé de déterminer de façon appropriée les taux de facteur VIII afin d’évaluer la dose à administrer ainsi que la fréquence du renouvellement des injections. Selon les patients, la réponse au facteur VIII peut varier, entraînant des taux de récupération et des demi-vies différents. Il peut être nécessaire d’ajuster les doses basées sur le poids corporel chez les patients présentant une insuffisance pondérale ou un surpoids. Dans le cas d’interventions chirurgicales majeures en particulier, un contrôle précis du traitement substitutif par une analyse de l’activité coagulante (activité plasmatique du facteur VIII) est indispensable.
Posologie
La dose et la durée du traitement substitutif dépendent de la sévérité du déficit en facteur VIII, de la localisation et de l'importance de l’épisode hémorragique, ainsi que de l'état clinique du patient.
Le nombre d’unités de facteur VIII administrées est exprimé en Unités Internationales (UI), conformément au standard actuel de l'OMS pour les concentrés de facteur VIII. L'activité coagulante en facteur VIII dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal), soit, de préférence, en Unités Internationales (UI) (conformément au Standard international du facteur VIII plasmatique).
Une Unité Internationale (UI) d'activité du facteur VIII correspond à la quantité de facteur VIII contenue dans un mL de plasma humain normal.
Traitement à la demande
Le calcul de la dose nécessaire de facteur VIII est basé sur le résultat empirique qu'une UI de facteur VIII par kg de poids corporel augmente l'activité coagulante plasmatique du facteur VIII de 2 UI/dL. La dose nécessaire est déterminée à l'aide de la formule suivante :
Nombre d'unités (UI) nécessaires = poids corporel (kg) x augmentation souhaitée du taux de facteur VIII (%) x 0,5.
La dose et la fréquence d’administration doivent être adaptées à l’efficacité clinique chez chaque individu. Dans certaines circonstances (par exemple, présence d’un inhibiteur à faible titre), des doses plus importantes que les quantités calculées à l’aide de la formule peuvent être nécessaires.
En cas de survenue de l'un des événements hémorragiques suivants, l'activité du facteur VIII ne doit pas chuter en dessous du taux d'activité plasmatique indiqué (en % de la normale ou UI/dL) pendant la période mentionnée. Le tableau ci‑dessous (tableau 1) peut servir de guide pour la détermination des posologies lors d'épisodes hémorragiques et de chirurgie :
Tableau 1 : Guide pour la détermination de la posologie lors d’épisodes hémorragiques et de chirurgie | ||
Degré de l'hémorragie / type d'intervention chirurgicale | Niveau de facteur VIII nécessaire (% ou UI/dL) | Fréquence des doses (heures) / durée du traitement (jours) |
Hémorragie |
|
|
Début d’hémarthrose, de saignement musculaire ou buccal. | 20 – 40 | Renouveler les injections toutes les 12 à 24 heures (toutes les 8 à 24 heures chez les patients âgés de moins de 6 ans) pendant au moins 1 jour, jusqu’à la fin de l’épisode hémorragique, indiquée par la disparition de la douleur ou l’obtention d’une cicatrisation. |
Hémarthrose plus étendue, hémorragie musculaire ou hématome. | 30 – 60 | Renouveler les injections toutes les 12 à 24 heures (toutes les 8 à 24 heures chez les patients âgés de moins de 6 ans) pendant 3 – 4 jours ou plus jusqu’à disparition de la douleur et de l’invalidité aiguë. |
Hémorragie mettant en jeu le pronostic vital. | 60 – 100 | Répéter les injections toutes les 8 à 24 heures (toutes les 6 à 12 heures chez les patients âgés de moins de 6 ans) jusqu’à disparition du risque vital. |
Chirurgie | 30 – 60 | |
Majeure | 80 – 100 | Renouveler les injections toutes les 8 à 24 heures (toutes les 6 à 24 heures chez les patients âgés de moins de 6 ans) jusqu’à cicatrisation satisfaisante de la plaie, puis poursuivre le traitement pendant au moins 7 jours supplémentaires pour maintenir une activité coagulante du facteur VIII entre 30 % et 60 % (UI/dL). |
Prophylaxie
Pour le traitement prophylactique à long terme des épisodes hémorragiques chez les patients atteints d’hémophilie A sévère, les posologies habituelles sont de 20 à 40 UI de facteur VIII par kg de poids corporel à des intervalles de 2 à 3 jours.
Dans certains cas, surtout chez le sujet jeune, des intervalles plus rapprochés ou des doses plus élevées peuvent être nécessaires.
Population pédiatrique
La posologie pour le traitement à la demande des patients pédiatriques (entre 0 et 18 ans) ne diffère pas de celle des patients adultes. Chez les patients de moins de 6 ans, les doses recommandées pour un traitement prophylactique sont de 20 à 50 UI de facteur VIII par kg de poids corporel 3 à 4 fois par semaine.
Mode d’administration
Voie intraveineuse. En cas d’administration par un non-professionnel de santé, une formation appropriée est nécessaire.
La vitesse d'administration sera déterminée en fonction du niveau de confort du patient et jusqu'à un maximum de 10 mL/min.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
Réaction allergique connue aux protéines de hamster ou de souris.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité
Des réactions d'hypersensibilité de type allergique, notamment l'anaphylaxie, ont été rapportées avec ADVATE. Le produit contient des traces de protéines de souris et de hamster. En cas de survenue de symptômes d’hypersensibilité, il faut indiquer aux patients d'interrompre immédiatement l'administration du produit et de contacter leur médecin. Les patients doivent être informés des signes précoces des réactions d’hypersensibilité, y compris de l’urticaire, de l’urticaire généralisé, de l’oppression thoracique, de la respiration sifflante, de l’hypotension et de l’anaphylaxie.
En cas de choc, le traitement standard relatif à l’état de choc devra être instauré.
En raison de la baisse de volume d‘injection d'ADVATE reconstitué dans 2 mL d'eau pour préparations injectables stérilisée, le temps de réaction et d'interruption de l‘injection en cas de réaction d'hypersensibilité est d'autant plus court. Il est donc conseillé de faire preuve de prudence lors de l‘injection d'ADVATE reconstitué dans 2 mL d'eau pour préparations injectables stérilisée, en particulier chez l'enfant.
Inhibiteurs
L'apparition d'anticorps neutralisants (inhibiteurs) du facteur VIII est une complication connue du traitement des patients atteints d'hémophilie A. Ces inhibiteurs sont habituellement des immunoglobulines IgG dirigées contre l’activité coagulante du facteur VIII et sont mesurés en Unités Bethesda (UB) par mL de plasma par le test modifié. Le risque de développer des inhibiteurs est corrélé à la gravité de la maladie ainsi qu’à l'exposition au facteur VIII, ce risque est le plus élevé au cours des 50 premiers jours d'exposition, mais il persiste tout au long de la vie bien qu’il soit peu fréquent.
La pertinence clinique de l’apparition d’inhibiteurs dépendra du titre d’inhibiteurs ; un faible titre présente un risque de réponse clinique insuffisante moins élevé qu’un titre élevé d’inhibiteurs.
De manière générale, tous les patients traités avec des produits de facteur VIII de coagulation doivent faire l’objet d’une surveillance soigneuse pour détecter l’apparition d'inhibiteurs par un suivi clinique et à l'aide de tests biologiques appropriés. Si le taux de facteur VIII plasmatique attendu n’est pas atteint ou si l’hémorragie n’est pas contrôlée par une dose adéquate, un dosage doit être réalisé afin de rechercher la présence d’un inhibiteur du facteur VIII. Chez les patients présentant un titre élevé d’inhibiteur, le traitement en facteur VIII peut ne pas être efficace et d’autres options thérapeutiques doivent être considérées. Le suivi de tels patients doit être effectué par des médecins expérimentés dans la prise en charge de l’hémophilie et des inhibiteurs du facteur VIII.
Administration incorrecte d'ADVATE
L'administration incorrecte (par voie intra-artérielle ou hors de la veine) d'ADVATE reconstitué avec 2 mL d'eau pour préparations injectables stérilisée peut entraîner des réactions transitoires légères au niveau du site d'injection, telles que des contusions et un érythème.
Événements cardiovasculaires
Chez les patients présentant des facteurs de risque cardiovasculaire, le traitement de substitution par facteur VIII peut accroître le risque cardiovasculaire.
Complications liées au cathéter
Si un dispositif d'accès veineux central (DAVC) est requis, le risque de complications liées au DAVC, notamment des infections locales, une bactériémie et une thrombose au site du cathéter, doit être pris en compte.
Considérations liées à l'excipient
Sodium
Ce médicament contient 10 mg de sodium par flacon, ce qui équivaut à 0,5 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
Il est fortement recommandé qu’à chaque administration d’ADVATE à un patient, le nom et le numéro de lot du produit soient enregistrés afin de maintenir un lien entre le patient et le numéro de lot du médicament.
Population pédiatrique
La liste des avertissements et des précautions s’applique aussi bien aux adultes qu’aux enfants.
Aucune interaction des produits à base de facteur VIII de coagulation humain (ADNr) avec d’autres médicaments n’a été rapportée.
Aucune étude de reproduction animale n‘a été conduite avec le facteur VIII. En raison de la rareté de l’hémophilie A chez la femme, il n’y a pas de donnée disponible sur l’utilisation de facteur VIII lors de la grossesse ou de l’allaitement. En conséquence, le facteur VIII ne doit être utilisé pendant la grossesse ou l’allaitement qu’en cas de nécessité absolue.
ADVATE n’a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Résumé du profil d'innocuité
Les études cliniques avec ADVATE comprenaient 418 sujets ayant connu au moins une exposition à ADVATE ; un total de 93 effets indésirables a été rapporté. Les effets indésirables qui ont été observés le plus fréquemment étaient le développement d’anticorps neutralisants du facteur VIII (inhibiteurs), des maux de tête et de la fièvre.
Une hypersensibilité ou des réactions allergiques (qui peuvent inclure : angioedème, brûlure et picotements au site de perfusion, frissons, rougeurs, urticaire généralisée, céphalées, urticaire, hypotension, léthargie, nausées, impatience, tachycardie, oppression thoracique, fourmillements, vomissements, sibilances) ont été rarement observées et peuvent, dans certains cas, évoluer vers une anaphylaxie sévère (y compris un choc).
L’apparition d’anticorps dirigés contre des protéines de souris et/ou de hamster peut être observée en rapport avec des réactions d’hypersensibilité.
Des anticorps neutralisants (inhibiteurs) peuvent apparaître chez des patients atteints d’hémophilie A traités avec le facteur VIII, y compris avec ADVATE (voir rubrique 5.1). Une telle apparition se manifeste par une réponse clinique insuffisante. Dans ce cas, il est recommandé de contacter un centre spécialisé en hémophilie.
Tableau de résumé des effets indésirables
Le tableau 2 présente la fréquence des effets indésirables issus des études cliniques et des signalements spontanés, conformément à la classification des systèmes d’organes MedDRA. (CSO et terme préconisé).
La fréquence a été évaluée selon les critères suivants : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/ 100), rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 : Fréquence des effets indésirables issus des études cliniques et des signalements spontanés | ||
Norme MedDRA | Effets indésirables | Fréquence des effets indésirablesa |
Infections et infestations | Grippe | Peu fréquent |
Laryngite | Peu fréquent | |
Affections hématologiques et du système lymphatique | Inhibition du facteur VIII | Peu fréquent (PPT)b |
Lymphangite | Peu fréquent | |
Affections du système immunitaire | Réaction anaphylactique* | Fréquence indéterminée |
Hypersensibilitéc* | Fréquence indéterminée | |
Affection du système nerveux | Maux de tête | Fréquent |
Vertiges | Peu fréquent | |
Troubles de la mémoire | Peu fréquent | |
Syncope | Peu fréquent | |
Tremblements | Peu fréquent | |
Migraine | Peu fréquent | |
Dysgeusie | Peu fréquent | |
Affections oculaires | Inflammation oculaire | Peu fréquent |
Affections cardiaques | Palpitations | Peu fréquent |
Affections vasculaires | Hématomes | Peu fréquent |
Bouffées de chaleur | Peu fréquent | |
Pâleur | Peu fréquent | |
Affections respiratoires, thoraciques et médiastinales | Dyspnées | Peu fréquent |
Affections gastro‑intestinales | Diarrhées | Peu fréquent |
Douleur abdominale haute | Peu fréquent | |
Nausées | Peu fréquent | |
Vomissements | Peu fréquent | |
Affections de la peau et du tissu sous‑cutané | Prurit | Peu fréquent |
Rash | Peu fréquent | |
Hyperhidrose | Peu fréquent | |
Urticaire | Peu fréquent | |
Troubles généraux et anomalies au site d’administration | Pyrexie | Fréquent |
Œdème périphérique | Peu fréquent | |
Douleur thoracique | Peu fréquent | |
Inconfort thoracique | Peu fréquent | |
Frissons | Peu fréquent | |
Etat anormal | Peu fréquent | |
Hématome au site de ponction vasculaire | Peu fréquent | |
Fatigue* | Fréquence indéterminée | |
Réaction au site d’injection* | Fréquence indéterminée | |
Malaise* | Fréquence indéterminée | |
Investigations | Augmentation du nombre de monocytes | Peu fréquent |
Diminution du facteur VIII de coagulationd | Peu fréquent | |
Diminution de l’hématocrite | Peu fréquent | |
Test biologique anormal | Peu fréquent | |
Lésions, intoxications et complications liées aux procédures | Complication post‑procédure | Peu fréquent |
Hémorragie post‑procédure | Peu fréquent | |
Réaction sur le site de l’intervention | Peu fréquent | |
a) Calculé sur la base du total de patients ayant reçu ADVATE (418) dans les études cliniques, à l’exception des effets indésirables identifiés dans le cadre de la surveillance post-commercialisation, marqués d’un *.
b) La fréquence est déterminée d’après des études portant sur tousdLes produits facteur VIII menées auprès de patients atteints d’hémophilie A sévère. PPT = patients précédemment traités, PUP = patients non traités précédemment (previously-untreated patients).
c) Effet indésirable expliqué dans la section ci-dessous.
d) La diminution inattendue de l’activité coagulante du facteur VIII est survenue chez un patient sous perfusion continue d’ADVATE après une chirurgie (jours post‑opératoires 10‑14). L’hémostase a été maintenue à tout moment pendant cette période. L’activité du facteur VIII et la clairance sont revenues à la normale à J15. Les recherches d’inhibiteur de facteur VIII réalisées à l’arrêt de la perfusion continue et à la fin de l’étude se sont révélées négatives.
Description de certains effets indésirables
Effets indésirables spécifiques aux résidus du processus de fabrication
Sur les 229 patients traités, pour lesquels le taux d’anticorps dirigés contre les protéines de cellules d’ovaire de hamster Chinois (CHO) a été évalué, ont été observés 3 cas d’augmentation statistiquement significative du titre, 4 cas de pics prolongés ou provisoires et un patient qui a présenté les deux à la fois mais sans aucun symptôme clinique. Sur les 229 patients traités, pour lesquels les anticorps dirigés contre les IgG d’origine murine ont été évalués, ont été observés 10 cas d’augmentation statistiquement significative des anticorps dirigés contre les antigènes murins, 2 cas de pics prolongés ou provisoires et un patient qui a présenté les deux à la fois. Chez quatre de ces patients, des cas isolés d’urticaire, de prurit, de rash et de numérations éosinophiles légèrement élevées ont été constatés au cours des expositions répétées au produit pendant l’étude.
Hypersensibilité
Les réactions de type allergique englobent l'anaphylaxie et se sont manifestées sous forme de vertiges, paresthésies, rash, bouffées congestives, gonflement facial, urticaire et prurit.
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables chez les enfants devraient être les mêmes que chez les adultes.
Outre le développement d'inhibiteurs chez des patients pédiatriques non préalablement traités (PUPs), et les complications liées au cathéter, aucune différence d'effets indésirables liée à l'âge n'a été constatée dans les études cliniques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
Aucun cas de surdosage suite à l'administration de facteur VIII de coagulation recombinant n'a été signalé.
Classe pharmacothérapeutique : antihémorragique ; facteur VIII de coagulation sanguin.
Code ATC : B02BD02.
Mécanisme d’action
ADVATE contient du facteur VIII de coagulation recombinant (octocog alfa), une glycoprotéine biologiquement équivalente à la glycoprotéine du facteur VIII présente dans le plasma humain. L’octocog alfa est une glycoprotéine constituée de 2 332 acides aminés avec un poids moléculaire approximatif de 280 kD.
Le complexe facteur VIII/facteur von Willebrand se compose de deux molécules (facteur VIII et facteur von Willebrand) aux fonctions physiologiques différentes. Lorsqu’il est perfusé à un patient hémophile, le facteur VIII se lie dans la circulation sanguine au facteur von Willebrand endogène. Le facteur VIII activé agit comme cofacteur du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation d’un caillot. L’hémophilie A est une maladie de la coagulation sanguine, héréditaire liée au sexe, due à la diminution de l’activité du facteur VIII:C provoquant des accidents hémorragiques au niveau des articulations, des muscles ou des organes internes, soit spontanément, soit à la suite d’un traumastisme accidentel ou chirurgical.
Le taux plasmatique en facteur VIII est augmenté grâce au traitement substitutif, ce qui permet de corriger temporairement le déficit en facteur VIII et les tendances hémorragiques.
Il convient de noter que le taux de saignement annualisé (TSA) n’est pas comparable entre les différents concentrés de facteur et entre les différentes études cliniques.
Efficacité et sécurité cliniques
Des données ont été collectées chez des patients avec inhibiteurs traités par induction de tolérance immune (ITI). Dans le cadre d’une sous‑étude de l'étude PUP (étude 060103), des traitements par ITI ont été documentés chez 11 PUPs. Une analyse rétrospective a été menée chez 30 patients pédiatriques sous ITI (dans l’étude 060703). Un registre prospectif non interventionnel (PASS-INT-004) a documenté l’ITI chez 44 patients pédiatriques et adultes dont 36 ont terminé un traitement par ITI. Les données démontrent qu’une tolérance immune peut être obtenue.
Lors de l'étude 060201, deux schémas de traitement prophylactique à long terme ont été comparés chez 53 PPT : un schéma posologique personnalisé adapté aux propriétés pharmacocinétiques (entre 20 et 80 UI de facteur VIII par kg de poids corporel à intervalles de 72 ± 6 heures, n = 23) et un schéma posologique prophylactique standard (20 à 40 UI/kg toutes les 48 ± 6 heures, n = 30). Le schéma posologique adapté aux propriétés pharmacocinétiques (selon une formule spécifique) était ciblé de manière à maintenir des taux minimum de facteur VIII ≥ 1 % pendant l'intervalle de 72 heures entre deux doses. Les résultats de cette étude démontrent que les deux schémas posologiques prophylactiques sont comparables en termes de réduction du taux d'hémorragie.
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec ADVATE dans tous les sous-groupes de la population pédiatrique atteints d’hémophilie A (déficit congénital en facteur VIII) dans l’« Induction de Tolérance Immune (ITI) chez les patients souffrant d’hémophilie A (déficit congénital en facteur VIII) et ayant développé des inhibiteurs du facteur VIII » et « traitement et prophylaxie des hémorragies chez les patients souffrant d’hémophilie A (déficit congénital en facteur VIII) » (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Toutes les études pharmacocinétiques avec ADVATE ont été réalisées chez des hémophiles A sévères à modérément sévères (taux basal de facteur VIII ≤ 2 %) préalablement traités. L’analyse des échantillons de plasma a été effectuée par un laboratoire central à l’aide du test chronométrique en un temps.
Au total, 195 patients atteints d'hémophilie A sévère (taux basal de facteur VIII < 1 %) ont fourni des paramètres pharmacocinétiques (PK) inclus dans la série d'analyses pharmacocinétiques per protocole. Des catégories de ces analyses pour les nourrissons (1 mois à < 2 ans), les enfants (2 à < 5 ans), les enfants plus âgés (5 à < 12 ans), les adolescents (12 à < 18 ans) et les adultes (18 ans et plus) ont été utilisées pour résumer les paramètres pharmacocinétiques, l'âge utilisé comme critère étant l'âge du patient au moment de la perfusion.
Tableau 3 : Résumé des paramètres pharmacocinétiques concernant ADVATE par tranche d'âge chez des patients atteints d'hémophilie A sévère (taux basal de facteur VIII < 1 %) | |||||
Paramètre (moyenne ± écart-type) | Nourrissons | Enfants | Enfants plus âgés | Adolescents | Adultes |
AUC totale (UI*·h/dL) | 1 362,1 ± 311,8 | 1 180,0 ± 432,7 | 1 506,6 ± 530,0 | 1 317,1 ± 438,6 | 1 538,5 ± 519,1 |
Récupération incrémentielle corrigée à Cmax (UI/dL par UI/kg)a | 2,2 ± 0,6 | 1,8 ± 0,4 | 2,0 ± 0,5 | 2,1 ± 0,6 | 2,2 ± 0,6 |
Demi-vie (h) | 9,0 ± 1,5 | 9,6 ± 1,7 | 11,8 ± 3,8 | 12,1 ± 3,2 | 12,9 ± 4,3 |
Concentration plasmatique maximale après la perfusion (UI/dL) | 110,5 ± 30,2 | 90,8 ± 19,1 | 100,5 ± 25,6 | 107,6 ± 27,6 | 111,3 ± 27,1 |
Temps de séjour moyen (h) | 11,0 ± 2,8 | 12,0 ± 2,7 | 15,1 ± 4,7 | 15,0 ± 5,0 | 16,2 ± 6,1 |
Volume de distribution à l'état d'équilibre (dL/kg) | 0,4 ± 0,1 | 0,5 ± 0,1 | 0,5 ± 0,2 | 0,6 ± 0,2 | 0,5 ± 0,2 |
Clairance (mL/kg*h) | 3,9 ± 0,9 | 4,8 ± 1,5 | 3,8 ± 1,5 | 4,1 ± 1,0 | 3,6 ± 1,2 |
a) Calculée à l'aide de la formule (Cmax - taux basal de facteur VIII) divisée par la dose en UI/kg, où Cmax représente le taux maximal de facteur VIII après la perfusion.
Population pédiatrique
L'innocuité et l'efficacité hémostatique d'ADVATE dans la population pédiatrique sont similaires aux valeurs enregistrées chez les patients adultes. La récupération corrigée et la demi‑vie terminale sont environ 20 % inférieures chez les jeunes enfants (mois de 6 ans) à celle des adultes, ce qui serait en partie dû au volume de plasma par kg de poids corporel plus élevé chez les patients plus jeunes.
Il n’existe actuellement aucune donnée de pharmacocinétique disponible avec ADVATE sur des patients non précédemment traités.
Les données non cliniques issues des études de pharmacologie de sécurité, toxicologie aiguë, toxicité en administration répétée, toxicité locale et génotoxicité n’ont pas révélé de risque particulier pour l’homme.
Une étude de tolérance locale chez le lapin a démontré qu'ADVATE reconstitué avec 2 mL d'eau pour préparations injectables stérilisée était bien toléré après administration intraveineuse. Une légère rougeur transitoire au site d'administration a été observée après administration intra‑artérielle et après administration à proximité de la veine. Néanmoins, aucun changement histopathologique indésirable lié n'a pu être observé, ce qui corrobore la nature transitoire de ce résultat.
Poudre
Mannitol (E421)
Chlorure de sodium
Histidine
Tréhalose
Chlorure de calcium (E509)
Trométamol
Polysorbate 80 (E433)
Glutathion (réduit).
Solvant
Eau pour préparations injectables stérilisée.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Flacon non ouvert
Deux ans.
Pendant la durée de conservation, le produit peut être conservé à température ambiante (ne dépassant pas 25 °C) pendant une période unique de 6 mois maximum. La date de fin de la période de 6 mois de conservation à température ambiante doit être indiquée sur l’emballage. À la fin de cette période, le produit doit être utilisé ou éliminé. Le produit ne doit pas être remis au réfrigérateur.
Après reconstitution
Après reconstitution, d’un point de vue microbiologique, le produit doit être utilisé immédiatement.
Cependant, la stabilité chimique et physique en cours d’utilisation a été démontrée pendant 3 heures à 25 °C.
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
ADVATE avec le dispositif BAXJECT II : conserver le flacon de produit dans l’emballage extérieur à l’abri de la lumière.
ADVATE dans le système BAXJECT III : conserver la plaquette scellée dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation de la solution après reconstitution, voir la rubrique 6.3.
Le flacon de poudre et le flacon contenant 2 mL de solvant sont en verre de type I fermés par des bouchons en caoutchouc chlorobutyle ou bromobutyle. Le produit est fourni dans l'une des deux configurations suivantes :
- ADVATE avec le dispositif BAXJECT II : chaque coffret contient un flacon de poudre et un flacon de 2 mL de solvant. Chaque coffret contient un dispositif pour la reconstitution (BAXJECT II).
- ADVATE dans le système BAXJECT III : chaque coffret contient un système BAXJECT III prêt à l'emploi dans une plaquette scellée (le flacon de poudre et le flacon contenant 2 mL de solvant sont préassemblés avec le système pour reconstitution).
6.6 Précautions particulières pour l'élimination et la manipulation
ADVATE doit être administré par voie intraveineuse après reconstitution du produit.
Le médicament reconstitué doit être inspecté visuellement pour mettre en évidence la présence de particules et d'une coloration anormale avant l’administration.
La solution doit être limpide, incolore et exempte de particules. Ne pas utiliser de solution trouble ou présentant des dépôts.
- L’utilisation d’une seringue Luer‑Lock est recommandée pour l’administration.
- Après reconstitution, la préparation doit être utilisée dans les 3 heures.
- Ne pas réfrigérer la préparation après reconstitution.
- Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Reconstitution avec le dispositif BAXJECT II
- Pour la reconstitution, utiliser uniquement l’eau pour préparations injectables stérilisée et le nécessaire de reconstitution fournis dans le coffret.
- Ne pas utiliser si le dispositif BAXJECT II, l’opercule ou l’emballage est endommagé ou présente des signes de détérioration.
- Utiliser une technique aseptique.
1. Si le produit est encore stocké au réfrigérateur, sortir les flacons de poudre ADVATE (poudre) et de solvant du réfrigérateur et laisser les atteindre la température ambiante (entre 15 °C et 25 °C).
2. Lavez‑vous soigneusement les mains à l’eau chaude et au savon.
3. Retirer les opercules des flacons de poudre et de solvant.
4. Nettoyer les bouchons avec les tampons d’alcool. Disposer les flacons sur une surface plane et propre.
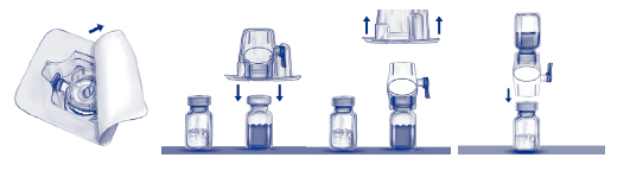
5. Ouvrir l’emballage de BAXJECT II en retirant le couvercle sans toucher l’intérieur (Fig. a). Ne pas retirer le dispositif de l’emballage. Ne pas utiliser si le dispositif BAXJECT II, l’opercule ou l’emballage est endommagé ou présente des signes de détérioration.
6. Retourner l’emballage et insérer le perforateur en plastique transparent dans le bouchon du flacon de solvant. Saisir l’emballage sur les côtés puis retirer l’emballage du dispositif BAXJECT II (Fig. b). Ne pas retirer le capuchon bleu du dispositif BAXJECT II.
7. Pour la reconstitution, n’utilisez que l’eau pour préparations injectables stérilisée et le dispositif médical fournis. En maintenant le dispositif BAXJECT II solidaire du flacon de solvant, tourner le système sur lui‑même de sorte que le flacon de solvant se trouve en haut. Insérer le perforateur en plastique blanc dans le bouchon du flacon de poudre ADVATE. Le vide entraînera le solvant vers le flacon de poudre ADVATE (Fig. c).
8. Agiter doucement jusqu’à ce que toute la poudre soit dissoute. Bien vérifier que la poudre ADVATE est complètement dissoute, sinon la totalité de la solution reconstituée ne passera pas au travers du filtre. Le produit se dissout rapidement (en général en moins d’une minute). Après reconstitution, la solution doit être limpide, incolore et exempte de particules.
Fig. a Fig. b Fig. c
Reconstitution avec le système BAXJECT III
- Ne pas utiliser si le couvercle n'est pas complètement scellé sur la plaquette
1. Si le produit est encore conservé au réfrigérateur, retirer la plaquette scellée (contient les flacons de solvant et de poudre préassemblés avec le système pour reconstitution) du réfrigérateur et la laisser revenir à température ambiante (entre 15 °C et 25 °C).
2. Se laver soigneusement les mains à l'eau chaude et au savon.
3. Ouvrir l'emballage ADVATE en décollant le couvercle. Retirer le système BAXJECT III de la plaquette.
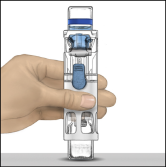
4. Placer ADVATE sur une surface plane avec le flacon de solvant en haut (Fig. 1). Le flacon de solvant porte une bande bleue. Ne retirer le capuchon bleu que lorsque vous serez invité à le faire, ultérieurement.
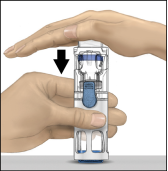
5. Tout en tenant ADVATE d'une main dans le système BAXJECT III, appuyer fermement sur le flacon de solvant de l'autre jusqu'à ce que le système soit entièrement replié et que le solvant s'écoule dans le flacon ADVATE (Fig. 2). N'incliner le système qu'une fois que le transfert est terminé.
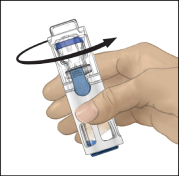
6. Vérifier que le transfert de solvant est terminé. Agiter doucement jusqu'à ce que toute la poudre soit dissoute (Fig. 3). Bien vérifier que la poudre ADVATE est complètement dissoute, sinon la totalité de la solution reconstituée ne passera pas au travers du filtre. Le produit se dissout rapidement (en général en moins de 1 minute). Après reconstitution, la solution doit être limpide, incolore et exempte de particules.
Fig. 1 | Fig. 2 | Fig. 3 |
|
|
|
Administration
Utiliser une technique aseptique.
Avant administration, la recherche de particules en suspension doit être réalisée sur tous les médicaments injectables quand la solution et le récipient le permettent. N’utiliser la solution que si elle est limpide et incolore.
1. Retirer l’opercule bleu du dispositif BAXJECT II/système BAXJECT III. Ne pas remplir la seringue d'air. Connecter la seringue au dispositif BAXJECT II/système BAXJECT III.
2. Retourner le système (le flacon contenant la solution reconstituée doit être désormais en position haute). Remplir la seringue avec la solution reconstituée en tirant lentement le piston en arrière.
3. Retirer la seringue.
4. Fixer l’aiguille à ailettes à la seringue. Injecter par voie intraveineuse. La solution devra être administrée lentement, à une vitesse tenant compte du confort du patient, et n’excédant pas 10 mL/minute. Le pouls doit être pris avant et pendant l'administration d’ADVATE. Si une augmentation importante du pouls apparaît, la diminution de la vitesse d'administration ou l'arrêt temporaire de l'injection permet généralement la disparition rapide des symptômes. (voir les paragraphes 4.4 et 4.8).
Takeda Manufacturing Austria AG
Industriestrasse, 67
A‑1221 Vienne
Autriche
medinfoEMEA@takeda.com
EU/1/03/271/007
EU/1/03/271/008
EU/1/03/271/009
EU/1/03/271/010
EU/1/03/271/017
EU/1/03/271/018
EU/1/03/271/019
EU/1/03/271/020
Date de première autorisation : 2 mars 2004
Date du dernier renouvellement : 20 décembre 2013
10. DATE DE MISE À JOUR DU TEXTE
05/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne du médicament https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 2955342 | ADVATE 2000UI PULV+SOLV SOL INJ 5 ML(400IU/ML)+KIT | B02BD02 | € 1327,95 | - | Oui | - | - |
| 2955359 | ADVATE 3000UI PULV+SOLV SOL INJ 5 ML(600IU/ML)+KIT | B02BD02 | € 1987,22 | - | Oui | - | - |
| 3342870 | ADVATE 250 UI PULV+SOLV SOL INJ 2 ML(125IU/ML)+KIT | B02BD02 | € 162,71 | - | Oui | € 2 | € 1 |
| 3342888 | ADVATE 500 UI PULV+SOLV SOL INJ 2 ML(250IU/ML)+KIT | B02BD02 | € 314,45 | - | Oui | € 2 | € 1 |
| 3342896 | ADVATE 1000UI PULV+SOLV SOL INJ 2 ML(500IU/ML)+KIT | B02BD02 | € 617,95 | - | Oui | € 2 | € 1 |
| 3342904 | ADVATE 1500UI PULV+SOLV SOL INJ 2 ML(750IU/ML)+KIT | B02BD02 | € 1003,26 | - | Oui | € 2 | € 1 |
| 3342912 | ADVATE 2000UI PULV+SOLV SOL INJ 5 ML(400IU/ML)+KIT | B02BD02 | € 1269,09 | - | Oui | € 2 | € 1 |
| 3342920 | ADVATE 3000UI PULV+SOLV SOL INJ 5 ML(600IU/ML)+KIT | B02BD02 | € 1952,9 | - | Oui | € 2 | € 1 |