1. DÉNOMINATION DU MÉDICAMENT
Tecfidera 120 mg, gélules gastro-résistantes
Tecfidera 240 mg, gélules gastro-résistantes
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Tecfidera 120 mg, gélules gastro-résistantes
Chaque gélule gastro-résistante contient 120 mg de diméthyl fumarate.
Tecfidera 240 mg, gélules gastro-résistantes
Chaque gélule gastro-résistante contient 240 mg de diméthyl fumarate.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Gélule gastro-résistante
Tecfidera 120 mg, gélules gastro-résistantes
Gélules gastro-résistantes vertes et blanches, de taille 0, portant l’inscription « BG-12 120 mg », contenant des micro-comprimés.
Tecfidera 240 mg, gélules gastro-résistantes
Gélules gastro-résistantes vertes, de taille 0, portant l’inscription « BG-12 240 mg », contenant des micro-comprimés.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Tecfidera est indiqué dans le traitement des adultes et des enfants âgés de 13 ans et plus atteints de sclérose en plaques de forme rémittente récurrente (SEP‑RR).
4.2 Posologie et mode d’administration
Le traitement doit être instauré sous la surveillance d’un médecin expérimenté dans la prise en charge de la sclérose en plaques.
Posologie
La dose initiale est de 120 mg deux fois par jour. Après 7 jours de traitement, la dose doit être augmentée à la dose d’entretien recommandée de 240 mg deux fois par jour (voir rubrique 4.4).
En cas d’oubli d’une dose, le patient ne doit pas prendre de dose double. Il ne peut prendre la dose oubliée qu’en respectant un intervalle de 4 heures entre les doses. Sinon, le patient doit attendre et prendre la dose suivante au moment habituel.
Une réduction temporaire de la dose à 120 mg deux fois par jour peut permettre de réduire la fréquence des bouffées congestives et des effets indésirables gastro-intestinaux. Il convient de revenir à la dose d’entretien recommandée de 240 mg deux fois par jour au cours du mois suivant.
Tecfidera doit être pris au moment des repas (voir rubrique 5.2). Chez les patients présentant des effets indésirables gastro-intestinaux ou des bouffées congestives, la prise de Tecfidera au moment des repas peut améliorer la tolérance (voir rubriques 4.4, 4.5 et 4.8).
Populations particulières
Sujets âgés
Les études cliniques réalisées avec Tecfidera ont concerné un nombre limité de patients âgés de 55 ans et plus ainsi qu’un nombre insuffisant de patients âgés de 65 ans et plus ce qui n’a pas permis de déterminer si cette population de patients répondait différemment à ce médicament par comparaison à des patients plus jeunes (voir rubrique 5.2). Compte tenu du mécanisme d’action de cette substance active, il n’y a théoriquement aucune raison de modifier la posologie chez le sujet âgé.
Insuffisants rénaux et hépatiques
Tecfidera n’a pas été étudié chez les patients insuffisants rénaux ou hépatiques. Selon les études de pharmacologie clinique, aucune adaptation posologique n’est nécessaire (voir rubrique 5.2). Le traitement de patients présentant une insuffisance rénale sévère ou hépatique sévère doit être instauré avec prudence (voir rubrique 4.4).
Population pédiatrique
La posologie est la même chez les adultes et les enfants âgés de 13 ans et plus.
Les données disponibles chez les enfants âgés de 10 à 12 ans sont limitées. Les données actuellement disponibles sont décrites aux rubriques 4.8 et 5.1 mais aucune recommandation sur la posologie ne peut être donnée.
La sécurité et l’efficacité de Tecfidera chez les enfants âgés de moins de 10 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale.
La gélule doit être avalée entière. Ne pas écraser, ouvrir, dissoudre, sucer ou mâcher la gélule ou son contenu car le pelliculage gastro-résistant des micro-comprimés évite les effets irritants sur le tractus gastro‑intestinal.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Leucoencéphalopathie multifocale progressive (LEMP) suspectée ou confirmée.
4.8 Effets indésirables
Synthèse du profil de sécurité
Les effets indésirables les plus fréquents sont les bouffées congestives (35 %) et les effets gastro-intestinaux (c’est-à-dire, diarrhées (14 %), nausées (12 %), douleurs abdominales (10 %), douleurs abdominales hautes (10 %)). Les bouffées congestives et les effets gastro-intestinaux ont tendance à survenir en début de traitement (principalement au cours du premier mois) et chez les patients présentant des bouffées congestives et troubles gastro‑intestinaux, ces troubles peuvent éventuellement continuer de manière intermittente pendant le traitement par Tecfidera. Les effets indésirables rapportés le plus fréquemment et ayant entraîné l’arrêt du traitement sont les bouffées congestives (3 %) et les effets gastro-intestinaux (4 %).
Dans le cadre des études cliniques de phases II et III contrôlées versus placebo et non contrôlées, 2 513 patients ont reçu Tecfidera pendant une durée allant jusqu’à 12 ans, avec une exposition globale au produit équivalente à 11 318 patient-années. Au total, 1 169 patients ont été traités par Tecfidera pendant au moins 5 ans et 426 patients pendant au moins 10 ans. L’expérience au cours des essais cliniques non contrôlés est comparable à celle des essais cliniques contrôlés contre placebo.
Liste tabulée des effets indésirables
Les effets indésirables rapportés au cours des études cliniques, des études de sécurité post-autorisation et des déclarations spontanées sont présentés dans le tableau ci-dessous.
Les effets indésirables sont présentés selon les termes préférentiels de la base de données MedDRA et les classes de systèmes d’organes. L’incidence des effets indésirables ci-dessous est exprimée en fonction des catégories suivantes :
- Très fréquent (≥ 1/10)
- Fréquent (≥ 1/100, < 1/10)
- Peu fréquent (≥ 1/1 000, < 1/100)
- Rare (≥ 1/10 000, < 1/1 000)
- Très rare (< 1/10 000)
- Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Base de données MedDRA des classes de systèmes d’organes | Effet indésirable | Catégorie de fréquence |
Infections et infestations | Gastro-entérite | Fréquent |
Leucoencéphalopathie multifocale progressive (LEMP) | Fréquence indéterminée | |
Zona | Fréquence indéterminée | |
Affections hématologiques et du système lymphatique | Lymphopénie | Fréquent |
Leucopénie | Fréquent | |
Thrombocytopénie | Peu fréquent | |
Affections du système immunitaire | Hypersensibilité | Peu fréquent |
Anaphylaxie | Fréquence indéterminée | |
Dyspnée | Fréquence indéterminée | |
Hypoxie | Fréquence indéterminée | |
Hypotension | Fréquence indéterminée | |
Angiœdème | Fréquence indéterminée | |
Affections du système nerveux | Sensation de brûlures | Fréquent |
Affections vasculaires | Bouffées congestives | Très fréquent |
Bouffées de chaleur | Fréquent | |
Affections respiratoires, thoraciques et médiastinales | Rhinorrhée | Fréquence indéterminée |
Affections gastro-intestinales | Diarrhées | Très fréquent |
Nausées | Très fréquent | |
Douleurs abdominales hautes | Très fréquent | |
Douleurs abdominales | Très fréquent | |
Vomissements | Fréquent | |
Dyspepsie | Fréquent | |
Gastrite | Fréquent | |
Troubles gastro-intestinaux | Fréquent | |
Affections hépatobiliaires | Augmentation de l’aspartate aminotransférase | Fréquent |
Augmentation de l’alanine aminotransférase | Fréquent | |
Atteinte hépatique médicamenteuse | Rare | |
Affections de la peau et du tissu sous-cutané | Prurit | Fréquent |
Rash | Fréquent | |
Érythème | Fréquent | |
Alopécie | Fréquent | |
Affections du rein et des voies urinaires | Protéinurie | Fréquent |
Troubles généraux et anomalies au site d’administration | Sensation de chaleur | Fréquent |
Investigations | Présence de cétones dans les urines | Très fréquent |
Présence d’albumine dans les urines | Fréquent | |
Diminution du nombre de globules blancs | Fréquent |
Description de certains effets indésirables
Bouffées congestives
Dans les études contre placebo, l’incidence des bouffées congestives (34 % versus 4 %) et des bouffées de chaleur (7 % versus 2 %) était respectivement plus élevée chez les patients traités par Tecfidera que chez ceux recevant le placebo. Les bouffées congestives étaient habituellement décrites comme des bouffées congestives ou de chaleur, mais elles pouvaient également comprendre d’autres effets (chaleur, rougeur, démangeaisons ou sensation de brûlure, par exemple). Les bouffées congestives tendaient à survenir en début de traitement (principalement pendant le premier mois) et chez les patients qui les présentaient, ces effets pouvaient se manifester de manière intermittente pendant tout le traitement par Tecfidera. Dans la majorité des cas, ces bouffées congestives étaient d’une sévérité légère à modérée. Au total, 3 % des patients traités par Tecfidera ont arrêté le traitement en raison de bouffées congestives. L’incidence des bouffées congestives graves pouvant se caractériser par un érythème généralisé, un rash et/ou un prurit, a été observée chez moins de 1 % des patients traités par Tecfidera (voir rubriques 4.2, 4.4 et 4.5).
Effets indésirables gastro-intestinaux
L’incidence des effets gastro-intestinaux (tels que diarrhées [14 % versus 10 %], nausées [12 % versus 9 %], douleurs abdominales hautes [10 % versus 6 %], douleurs abdominales [9 % versus 4 %], vomissements [8 % versus 5 %] et dyspepsie [5 % versus 3 %]) était respectivement plus élevée chez les patients traités par Tecfidera que chez les patients sous placebo. L’incidence des effets indésirables gastro-intestinaux était plus élevée en début de traitement (principalement durant le premier mois) et chez les patients présentant des troubles gastro‑intestinaux, ces troubles peuvent éventuellement continuer de manière intermittente pendant le traitement par Tecfidera. Pour la majorité des patients présentant des troubles gastro-intestinaux, ces derniers étaient légers ou modérés. Quatre pour cent (4 %) des patients traités par Tecfidera ont dû arrêter leur traitement à cause d’effets indésirables gastro-intestinaux. L’incidence des effets gastro-intestinaux graves, notamment des gastro-entérites et des gastrites, a été observée chez 1 % des patients traités par Tecfidera (voir rubrique 4.2).
Fonction hépatique
Sur la base des données des études contrôlées contre placebo, chez la majorité des patients présentant des augmentations des transaminases hépatiques, ces augmentations étaient < 3 fois la LSN. L’incidence accrue d’une augmentation du taux des transaminases hépatiques chez les patients traités par Tecfidera, en comparaison au placebo, était principalement observée durant les 6 premiers mois de traitement. Une augmentation du taux d’alanine aminotransférase et d’aspartate aminotransférase ≥ 3 fois la LSN a été observée respectivement chez 5 % et 2 % des patients sous placebo et chez 6 % et 2 % des patients traités par Tecfidera. Les arrêts de traitement dus à un taux élevé de transaminases hépatiques ont été < 1 % et comparables chez les patients traités par Tecfidera et chez ceux sous placebo. Il n’a pas été observé d’augmentations des taux de transaminases ≥ 3 fois la LSN accompagnées d’augmentations du taux de bilirubine totale > 2 fois la LSN dans les études contrôlées contre placebo.
Des cas d’augmentation des enzymes hépatiques et d’atteinte hépatique médicamenteuse (élévations des transaminases ≥ 3 fois la LSN accompagnées d’élévations de la bilirubine totale > 2 fois la LSN) après l’administration de Tecfidera ont été rapportés depuis la commercialisation ; ils se sont résolus après l’arrêt du traitement.
Lymphopénie
Dans les études contrôlées contre placebo, la majorité des patients (> 98 %) présentait avant l’instauration du traitement des taux normaux de lymphocytes. Après le traitement par Tecfidera, le nombre moyen de lymphocytes a diminué au cours de la première année puis a atteint un plateau. En moyenne, le nombre de lymphocytes a diminué d’environ 30 % par rapport à la valeur initiale. Les nombres moyen et médian de lymphocytes sont restés dans les limites de la normale. Un nombre de lymphocytes < 0,5 × 109/L a été observé chez < 1 % des patients sous placebo et chez 6 % de ceux traités par Tecfidera. Un nombre de lymphocytes < 0,2 × 109/L a été observé chez 1 patient traité par Tecfidera contre aucun patient sous placebo.
Dans les études cliniques (contrôlées et non contrôlées), 41 % des patients traités par Tecfidera présentaient une lymphopénie (définie dans ces études comme < 0,91 × 109/L). Une lymphopénie légère (taux ≥ 0,8 × 109/L et < 0,91 × 109/L) a été observée chez 28 % des patients ; une lymphopénie modérée (taux ≥ 0,5 × 109/L et < 0,8 × 109/L) persistant pendant au moins six mois a été observée chez 11 % des patients ; une lymphopénie sévère (taux < 0,5 × 109/L) persistant pendant au moins six mois a été observée chez 2 % des patients. Dans le groupe présentant une lymphopénie sévère, les taux de lymphocytes sont restés < 0,5 × 109/L avec la poursuite du traitement chez la majorité des patients.
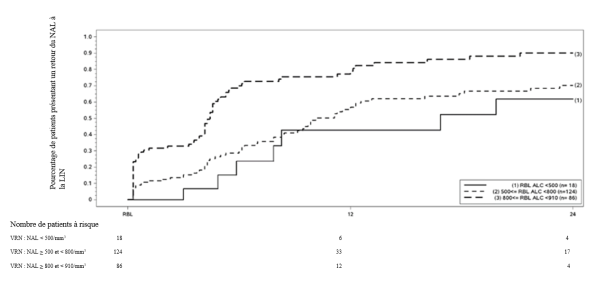
De plus, dans une étude prospective non contrôlée, réalisée après commercialisation, à la semaine 48 du traitement par Tecfidera (n = 185), le nombre de lymphocytes T CD4+ avait modérément (taux ≥ 0,2 × 109/L à < 0,4 × 109/L) ou sévèrement (< 0,2 × 109/L) diminué chez respectivement, 37 % ou 6 % des patients, tandis que les lymphocytes T CD8+ étaient plus fréquemment réduits, avec jusqu’à 59 % des patients ayant un taux < 0,2 × 109/L et 25 % des patients ayant un taux < 0,1 × 109/L. Dans les études cliniques contrôlées et non contrôlées, les patients qui arrêtaient le traitement par Tecfidera avec un taux de lymphocytes inférieur à la LIN étaient suivis afin de surveiller le retour à la normale (voir rubrique 5.1).
Leucoencéphalopathie multifocale progressive (LEMP)
Des cas d’infections par le virus de John Cunningham (JCV) provoquant une LEMP ont été rapportés avec Tecfidera (voir rubrique 4.4). La LEMP peut avoir une issue fatale ou entraîner un handicap sévère. Dans l’un des essais cliniques, 1 patient prenant Tecfidera a développé une LEMP dans le cadre d’une lymphopénie sévère et prolongée (nombre de lymphocytes principalement < 0,5 × 109/L pendant 3,5 ans), avec une issue fatale. Dans le cadre de la post-commercialisation, la LEMP est également survenue en présence d’une lymphopénie modérée et légère (> 0,5 × 109/L à < LIN, telle que définie par l’intervalle de référence du laboratoire local).
Dans plusieurs cas de LEMP avec détermination des sous-types de lymphocytes T au moment du diagnostic de la LEMP, on a constaté que le nombre de lymphocytes T CD8+ était réduit à < 0,1 × 109/L, alors que les réductions du nombre de lymphocytes T CD4+ étaient variables (allant de < 0,05 à 0,5 × 109/L) et étaient davantage corrélées avec la sévérité globale de la lymphopénie (< 0,5 × 109/L à < LIN). En conséquence, le rapport CD4+/CD8+ a augmenté chez ces patients.
Une lymphopénie modérée à sévère prolongée semble augmenter le risque de LEMP avec Tecfidera. Cependant, la LEMP est également survenue chez des patients présentant une lymphopénie légère. En outre, la majorité des cas de LEMP dans le cadre de la post-commercialisation sont survenus chez des patients > 50 ans.
Infections zostériennes
Des infections zostériennes (zona) ont été rapportées lors de l’utilisation de Tecfidera. Dans l’étude d’extension à long terme au cours de laquelle 1 736 patients atteints de SEP ont été traités, 5 % des patients ont présenté un ou plusieurs événements de type zona, dont 42 % étaient d’intensité légère, 55 % d’intensité modérée et 3 % d’intensité sévère. Le délai d’apparition allait d’environ 3 mois à 10 ans après l’administration de la première dose de Tecfidera. Quatre patients ont présenté des événements graves, qui se sont tous résolus. La plupart des patients, notamment ceux ayant présenté une infection zostérienne grave, avaient un nombre de lymphocytes supérieur à la limite inférieure de la normale. Chez une majorité de sujets dont le nombre de lymphocytes concomitant était inférieur à la LIN, la lymphopénie a été jugée modérée ou sévère. Depuis la commercialisation, la plupart des cas d’infection zostérienne (zona) étaient sans gravité et ont disparu après traitement. Les données disponibles concernant le nombre absolu de lymphocytes (NAL) chez les patients atteints d’infection herpétique depuis la commercialisation sont limitées. Toutefois, la plupart des patients chez qui le NAL a été rapporté ont présenté une lymphopénie modérée (≥ 0,5 × 109/L à < 0,8 × 109/L) ou sévère (< 0,5 × 109/L à 0,2 × 109/L) (voir rubrique 4.4).
Anomalies biologiques
Dans les études contrôlées contre placebo, le taux de cétones urinaires (1+ ou plus) était plus élevé chez les patients traités par Tecfidera (45 %) que chez ceux sous placebo (10 %), sans qu’aucune conséquence clinique négative n’ait été observée pendant les essais cliniques.
Le taux de 1,25-dihydroxy-vitamine D a diminué chez les patients traités par Tecfidera, comparé à ceux sous placebo (diminution du pourcentage médian, par rapport au pourcentage initial, à 2 ans respectivement de 25 % versus 15 %), alors que le taux d’hormone parathyroïdienne (PTH) a augmenté chez les patients traités par Tecfidera, par rapport à ceux sous placebo (augmentation du pourcentage médian, par rapport au pourcentage initial, à 2 ans respectivement de 29 % versus 15 %). Les valeurs moyennes de ces deux paramètres sont restées dans les limites de la normale.
Une augmentation transitoire du nombre moyen d’éosinophiles a été observée durant les deux premiers mois de traitement.
Population pédiatrique
Dans une étude en ouvert randomisée, contrôlée versus comparateur actif d’une durée de 96 semaines, des enfants et des adolescents atteints de SEP-RR (n = 7 âgés de 10 à moins de 13 ans et n = 71 âgés de 13 à moins de 18 ans) ont été traités à la dose de 120 mg deux fois par jour pendant 7 jours puis 240 mg deux fois par jour pendant le reste de la période de traitement. Le profil de sécurité chez ces patients était comparable à celui précédemment observé chez les patients adultes.
Le plan expérimental de l’étude clinique pédiatrique était différent de celui des études cliniques contrôlées versus placebo menées chez des adultes. Par conséquent, une contribution du plan expérimental de l’étude aux différences numériques des événements indésirables entre les populations pédiatrique et adulte ne peut être exclue. Des affections gastrointestinales ainsi que des affections respiratoires, thoraciques et médiastinales et des événements indésirables tels que céphalées et dysménorrhée ont été rapportés plus fréquemment (fréquence (≥ 10 %) dans la population pédiatrique que dans la population adulte. Les taux de ces événements indésirables rapportés chez les patients pédiatriques étaient les suivants :
- Des céphalées ont été rapportées chez 28 % des patients traités par Tecfidera versus 36 % des patients traités par l’interféron bêta‑1a.
- Des affections gastro‑intestinales ont été rapportées chez 74 % des patients traités par Tecfidera versus 31 % des patients traités par l’interféron bêta‑1a. Parmi celles‑ci, les plus fréquemment rapportées avec Tecfidera étaient des douleurs abdominales et des vomissements.
- Des affections respiratoires, thoraciques et médiastinales ont été rapportées chez 32 % des patients traités par Tecfidera versus 11 % des patients traités par l’interféron bêta‑1a. Parmi celles‑ci, les plus fréquemment rapportées avec Tecfidera étaient des douleurs oropharyngées et une toux.
- Des dysménorrhées ont été rapportées chez 17 % des patientes traitées par Tecfidera versus 7 % des patientes traitées par l’interféron bêta‑1a.
Dans une petite étude en ouvert non contrôlée d’une durée de 24 semaines menée chez des enfants et des adolescents atteints de SEP‑RR âgés de 13 à 17 ans (dose de 120 mg deux fois par jour pendant 7 jours puis 240 mg deux fois par jour pendant le reste de la période de traitement, n = 22), suivie d’une étude d’extension de 96 semaines (dose de 240 mg deux fois par jour, n = 20), le profil de sécurité était comparable à celui observé chez les patients adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
France
Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance
Site internet: https://signalement.social-sante.gouv.fr/
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Biogen Netherlands B.V.
Prins Mauritslaan 13
1171 LP Badhoevedorp
Pays-Bas
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/13/837/001
EU/1/13/837/002
EU/1/13/837/003
10. DATE DE MISE À JOUR DU TEXTE
12/2024
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3236080 | TECFIDERA 120MG GASTRO RESIST CAPS 14 | L04AX07 | € 98,73 | - | Oui | € 12,8 | € 8,5 |
| 3236106 | TECFIDERA 240MG GASTRO RESIST CAPS 56 | L04AX07 | € 362 | - | Oui | € 12,8 | € 8,5 |