RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Olumiant 1 mg comprimés pelliculés
Olumiant 2 mg comprimés pelliculés
Olumiant 4 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Olumiant 1 mg comprimés pelliculés
Chaque comprimé pelliculé contient 1 mg de baricitinib.
Olumiant 2 mg comprimés pelliculés
Chaque comprimé pelliculé contient 2 mg de baricitinib.
Olumiant 4 mg comprimés pelliculés
Chaque comprimé pelliculé contient 4 mg de baricitinib.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé)
Olumiant 1 mg comprimés pelliculés
Comprimé rond de 6,75 mm, de couleur rose très pâle, avec la mention « Lilly » gravée sur une face et le chiffre « 1 » gravé sur l’autre face.
Olumiant 2 mg comprimés pelliculés
Comprimé oblong de 9 x 7,5 mm, de couleur rose pâle, avec la mention « Lilly » gravée sur une face et le chiffre « 2 » gravé sur l’autre face.
Olumiant 4 mg comprimés pelliculés
Comprimé rond de 8,5 mm, de couleur rose moyen, avec la mention « Lilly » gravée sur une face et le chiffre « 4 » gravé sur l’autre face.
Le comprimé est doté d’une zone en creux de chaque côté.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Polyarthrite rhumatoïde
Le baricitinib est indiqué dans le traitement de la polyarthrite rhumatoïde active modérée à sévère chez les patients adultes qui ont présenté une réponse inadéquate, ou une intolérance, à un ou plusieurs traitements de fond (DMARDs). Le baricitinib peut être utilisé en monothérapie ou en association avec le méthotrexate (voir rubriques 4.4, 4.5 et 5.1 pour les données sur les différentes associations).
Dermatite atopique
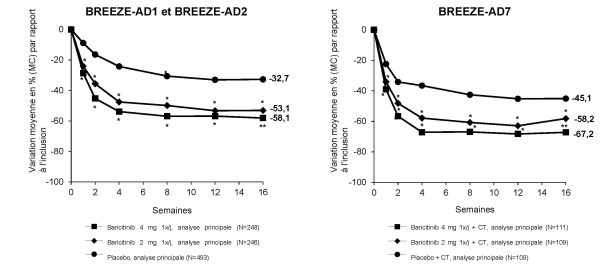
Le baricitinib est indiqué dans le traitement de la dermatite atopique modérée à sévère des patients adultes et pédiatriques âgés de 2 ans et plus qui nécessitent un traitement systémique.
Pelade (Alopecia areata)
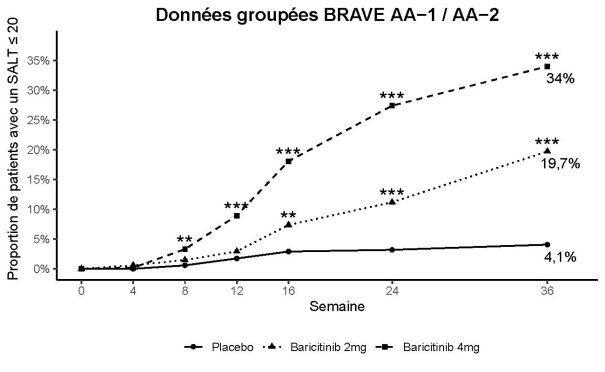
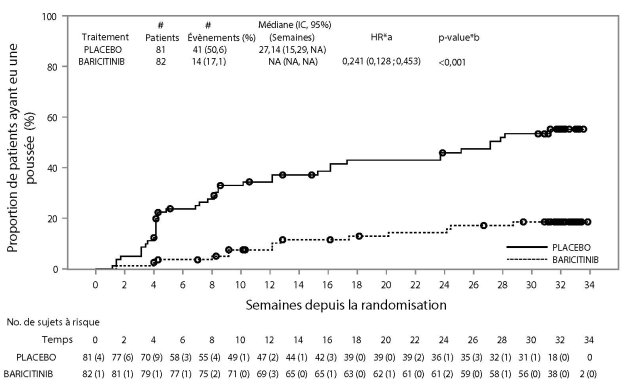
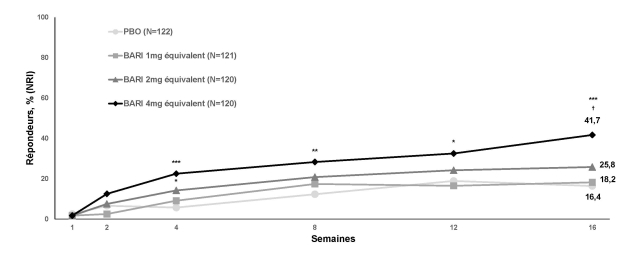
Le baricitinib est indiqué dans le traitement de la pelade sévère de l’adulte et de l’adolescent âgé de 12 ans et plus (voir rubrique 5.1)
Arthrite juvénile idiopathique
Le baricitinib est indiqué dans le traitement de l’arthrite juvénile idiopathique active chez les patients âgés de 2 ans et plus, qui ont présenté une réponse inadéquate ou une intolérance à un ou plusieurs traitements de fond (DMARDs) conventionnels synthétiques ou biologiques antérieurs :
- Arthrite juvénile idiopathique polyarticulaire (polyarthrite à facteur rhumatoïde positif [RF+] ou négatif [RF-], et oligoarthrite étendue),
- Arthrite liée à l’enthésite, et
- Rhumatisme psoriasique juvénile.
Le baricitinib peut être utilisé en monothérapie ou en association avec le méthotrexate.
4.2 Posologie et mode d’administration
Le traitement doit être initié par un médecin expérimenté dans le diagnostic et le traitement des pathologies pour lesquelles ce médicament est indiqué.
Posologie
Polyarthrite rhumatoïde
La dose recommandée de baricitinib est de 4 mg une fois par jour. Une dose de 2 mg une fois par jour est recommandée pour les patients à plus haut risque de thrombo-embolie veineuse (TEV), d’évènements indésirables cardiovasculaires majeurs (MACE) et de tumeurs malignes, pour les patients âgés de 65 ans et plus et pour les patients ayant des antécédents d’infections chroniques ou récurrentes (voir rubrique 4.4). Une dose de 4 mg une fois par jour peut être envisagée pour les patients dont l’activité de la maladie est insuffisamment contrôlée avec la dose de 2 mg une fois par jour. Une dose de 2 mg une fois par jour doit être envisagée pour les patients dont l’activité de la maladie est contrôlée durablement avec la dose de 4 mg une fois par jour et qui sont éligibles à une diminution de la dose (voir rubrique 5.1).
Dermatite atopique
Adultes
La dose recommandée de baricitinib est de 4 mg une fois par jour. Une dose de 2 mg une fois par jour est recommandée pour les patients à plus haut risque de thrombo-embolie veineuse (TEV), d’évènements indésirables cardiovasculaires majeurs (MACE) et de tumeurs malignes, pour les patients âgés de 65 ans et plus et pour les patients ayant des antécédents d’infections chroniques ou récurrentes (voir rubrique 4.4). Une dose de 4 mg une fois par jour peut être envisagée pour les patients dont l’activité de la maladie est insuffisamment contrôlée avec la dose de 2 mg une fois par jour. Une dose de 2 mg une fois par jour doit être envisagée pour les patients dont l’activité de la maladie est contrôlée durablement avec la dose de 4 mg une fois par jour et qui sont éligibles à une diminution de la dose (voir rubrique 5.1).
Le baricitinib peut être utilisé avec ou sans corticoïdes topiques. L’efficacité du baricitinib peut être améliorée lorsqu’associé aux corticoïdes topiques (voir rubrique 5.1). Il est possible d’utiliser des inhibiteurs topiques de la calcineurine, mais ils doivent être réservés aux zones sensibles uniquement, telles que le visage, le cou, les zones intertrigineuses et les parties génitales.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 8 semaines de traitement.
Enfants et adolescents (2 ans et plus)
La dose recommandée de baricitinib est de 4 mg une fois par jour pour les patients pesant 30 kg ou plus. Chez les patients pesant entre 10 kg et moins de 30 kg, la dose recommandée est de 2 mg une fois par jour. Une dose réduite de moitié doit être envisagée pour les patients dont l’activité de la maladie est contrôlée durablement avec la dose recommandée et qui sont éligibles à une diminution de la dose.
Le baricitinib peut être utilisé avec ou sans corticoïdes topiques. Il est possible d’utiliser des inhibiteurs topiques de la calcineurine, mais ils doivent être réservés aux zones sensibles uniquement, telles que le visage, le cou, les zones intertrigineuses et les parties génitales.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 8 semaines de traitement.
Pelade (Alopecia areata)
Adultes
La dose recommandée de baricitinib est de 4 mg une fois par jour. Une dose de 2 mg une fois par jour est recommandée pour les patients à plus haut risque de thrombo-embolie veineuse (TEV), d’évènements indésirables cardiovasculaires majeurs (MACE) et de tumeurs malignes, pour les patients âgés de 65 ans et plus et pour les patients ayant des antécédents d’infections chroniques ou récurrentes (voir rubrique 4.4). Une dose de 4 mg une fois par jour peut être envisagée pour les patients dont l’activité de la maladie est insuffisamment contrôlée avec la dose de 2 mg une fois par jour. Une dose de 2 mg une fois par jour doit être envisagée pour les patients dont l’activité de la maladie est contrôlée durablement avec la dose de 4 mg une fois par jour et qui sont éligibles à une diminution de la dose (voir rubrique 5.1).
Lorsqu'une réponse stable a été obtenue, il est recommandé de poursuivre le traitement pendant au moins plusieurs mois, afin d'éviter une rechute. Le rapport bénéfice/risque du traitement doit être réévalué à intervalles réguliers pour chaque patient.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 36 semaines de traitement.
Adolescents (âgés de 12 ans et plus)
La dose recommandée de baricitinib est de 4 mg une fois par jour pour les patients pesant 30 kg ou plus. Voir la rubrique 4.2 Population pédiatrique ci-dessous pour les informations sur les patients pesant moins de 30 kg. Une dose de 2 mg une fois par jour doit être envisagée pour les patients dont l’activité de la maladie est contrôlée durablement avec la dose de 4 mg une fois par jour et qui sont éligibles à une diminution de la dose.
Lorsqu’une réponse stable a été obtenue, il est recommandé de poursuivre le traitement pendant au moins plusieurs mois, afin d’éviter une rechute. Le rapport bénéfice/risque du traitement doit être réévalué à intervalles réguliers pour chaque patient.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 36 semaines de traitement.
Arthrite juvénile idiopathique (de 2 ans à moins de 18 ans)
La dose recommandée de baricitinib est de 4 mg une fois par jour pour les patients pesant 30 kg ou plus. Chez les patients pesant entre 10 kg et moins de 30 kg, la dose recommandée est de 2 mg une fois par jour.
L’arrêt du traitement doit être envisagé chez les patients qui ne présentent pas de bénéfice thérapeutique après 12 semaines de traitement.
Initiation du traitement
Le traitement ne doit pas être instauré chez des patients ayant un nombre absolu de lymphocytes inférieur à 0,5 x 109 cellules/L, un nombre absolu de polynucléaires neutrophiles inférieur à 1 x 109 cellules/L, ou un taux d’hémoglobine inférieur à 8 g/dL. Le traitement peut être instauré une fois que ces valeurs sont au-dessus de ces limites (voir rubrique 4.4).
Réduction de dose
Chez les patients traités par des inhibiteurs puissants du transporteur d’anion organique de type 3 (OAT3) comme le probénécide, ou ayant une clairance de la créatinine comprise entre 30 et 60 mL/min, la dose recommandée doit être réduite de moitié pour les patients pédiatriques et est de 2 mg pour les patients adultes (voir rubrique 4.5).
Populations particulières
Insuffisance rénale
Chez les patients adultes ayant une clairance de la créatinine comprise entre 30 et 60 mL/min, la dose recommandée est de 2 mg une fois par jour. Chez les patients pédiatriques ayant une clairance de la créatinine comprise entre 30 et 60 mL/min, la dose recommandée de baricitinib doit être réduite de moitié. L’administration de baricitinib n’est pas recommandée chez les patients ayant une clairance de la créatinine inférieure à 30 mL/min (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement de dose n’est nécessaire chez les patients ayant une insuffisance hépatique légère à modérée. L’administration de baricitinib n’est pas recommandée chez les patients présentant une insuffisance hépatique sévère (voir rubrique 5.2).
Personnes âgées
L’expérience clinique chez les patients âgés de 75 ans et plus est très limitée.
Population pédiatrique
La sécurité et l’efficacité du baricitinib chez les enfants âgés de moins de 2 ans ayant une dermatite atopique ou une arthrite juvénile idiopathique n’ont pas encore été établies. Aucune donnée n’est disponible. Voir rubrique 4.2 ci-dessus pour les informations de posologie chez les enfants âgés de 2 ans et plus.
La sécurité et l’efficacité du baricitinib chez les enfants de moins de 12 ans ou pesant < 30 kg ayant une pelade (alopecia areata) n’ont pas encore été établies. Aucune donnée n’est disponible. Voir rubrique 4.2 ci-dessus pour les informations de posologie chez les adolescents âgés de 12 ans et plus, et pesant 30 kg ou plus.
Chez les patients pédiatriques qui ne sont pas en mesure d’avaler des comprimés, les comprimés peuvent être dispersés dans de l’eau avant administration (voir ci-dessous « Autre mode d’administration pour les enfants »). La présentation suspension buvable peut également être utilisée si elle est disponible.
Mode d’administration
Voie orale.
Le baricitinib doit être pris une fois par jour avec ou sans aliments et peut être pris à tout moment de la journée.
Autre mode d’administration pour les enfants
Chez les patients pédiatriques qui ne sont pas en mesure d'avaler des comprimés entiers, il peut être envisagé de disperser les comprimés dans de l'eau. Seule l'eau doit être utilisée pour disperser le comprimé. Seul le nombre de comprimés nécessaires pour la dose doit être dispersé.
Si pour une raison quelconque la totalité de la suspension n'est pas administrée, ne pas disperser et administrer un autre comprimé, mais attendre la prochaine dose prévue.
Pour les instructions concernant la dispersion du médicament avant l'administration, voir rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Grossesse (voir rubrique 4.6).
4.8 Effets indésirables
Résumé du profil de tolérance
Les effets indésirables les plus fréquemment rapportés avec le baricitinib sont l’augmentation du LDL-cholestérol (26,0 %), les infections des voies respiratoires supérieures (16,9 %), les céphalées (5,2 %), l’Herpes simplex (3,2 %) et les infections des voies urinaires (2,9 %). Des cas de pneumonies graves (0,2 %) et de zonas graves (0,2 %) sont survenus de façon peu fréquente chez des patients atteints de polyarthrite rhumatoïde.
Liste tabulée des effets indésirables
Fréquence estimée : Très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000). Les fréquences présentées dans le Tableau 2 reposent sur l’intégration des données issues des études cliniques chez les adultes et/ou de l’expérience post-commercialisation pour les indications polyarthrite rhumatoïde, dermatite atopique et pelade (Alopecia areata), sauf mention contraire. Lorsque des différences notables de fréquence ont été observées entre les indications, elles sont présentées dans les notes sous le tableau. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2. Effets indésirables
Classe de systèmes d’organes | Très fréquent | Fréquent | Peu fréquent |
Infections et infestations | Infections des voies respiratoires supérieures | Pneumonied |
|
Affections hématologiques et du système lymphatique |
| Thrombocytose > 600 x 109 cellules/La, d | Neutropénie < 1 x 109 cellules/La |
Affections du système immunitaire |
|
| Gonflement du visage, Urticaire |
Troubles du métabolisme et de la nutrition | Hypercholestérolémiea |
| Hypertriglycéridémiea |
Affections du système nerveux |
| Céphalées |
|
Affections vasculaires |
|
| Thrombose veineuse profondeb |
Affections respiratoires, thoraciques et médiastinales |
|
| Embolie pulmonairef |
Affections gastro-intestinales |
| Douleurs abdominalesd | Diverticulite |
Affections hépatobiliaires |
| Élévation de l’ALAT ≥ 3 x LSNa, d | Élévation de l’ASAT ≥ 3 x LSNa, e |
Affections de la peau et du tissu sous-cutané |
| Éruption cutanée |
|
Investigations |
| Élévation de la créatine phosphokinase > 5 x LSNa, c | Prise de poids |
a Inclut les variations biologiques détectées pendant la surveillance réalisée en routine (voir texte ci-dessous).
b La fréquence des zona et thrombose veineuse profonde est basée sur les études cliniques portant sur la polyarthrite rhumatoïde.
c Dans les études cliniques portant sur la polyarthrite rhumatoïde, l’acné et l’élévation de la créatine phosphokinase > 5 x LSN ont été peu fréquentes.
d Dans les études cliniques portant sur la dermatite atopique, les nausées et l’élévation de l’ALAT ≥ 3 x LSN ont été peu fréquentes. Dans les études cliniques portant sur la pelade, les douleurs abdominales ont été peu fréquentes. Dans les études cliniques portant sur la dermatite atopique et la pelade, la pneumonie et la thrombocytose > 600 x 109 cellules/L ont été peu fréquentes.
e Dans les études cliniques portant sur la pelade, l’élévation de l’ASAT ≥ 3 x LSN a été fréquente.
f La fréquence de l’embolie pulmonaire est basée sur les études cliniques portant sur la polyarthrite rhumatoïde et la dermatite atopique.
g Les folliculites ont été observées dans les études cliniques portant sur la pelade. Elles étaient généralement localisées dans la zone du cuir chevelu associée à la repousse des cheveux.
Description d’effets indésirables sélectionnés
Affections gastro-intestinales
Pendant 52 semaines, chez les patients naïfs de tout traitement des études cliniques portant sur la polyarthrite rhumatoïde, la fréquence des nausées a été plus élevée avec l’association du méthotrexate avec le baricitinib (9,3 %) qu’avec le méthotrexate seul (6,2 %) ou le baricitinib seul (4,4 %). Dans les données intégrant les études cliniques portant sur la polyarthrite rhumatoïde, la dermatite atopique et la pelade, les nausées ont été plus fréquentes pendant les 2 premières semaines de traitement.
Les cas de douleurs abdominales ont été généralement d’intensité légère et étaient transitoires. Ils n’ont pas été associés à des troubles gastro-intestinaux infectieux ou inflammatoires et n’ont pas entraîné d’interruption du traitement.
Infections
Dans les données intégrant les études cliniques portant sur la polyarthrite rhumatoïde, la dermatite atopique et la pelade, la plupart des infections étaient d’intensité légère à modérée. Dans les études incluant les deux doses, des infections ont été rapportées chez 31,0 %, 25,7 % et 26,7 % des patients dans les groupes 4 mg, 2 mg et placebo, respectivement. Dans les études cliniques portant sur la polyarthrite rhumatoïde, l’association avec le méthotrexate a entraîné une augmentation de la fréquence des infections par rapport au traitement par baricitinib en monothérapie. Le zona a été fréquent dans la polyarthrite rhumatoïde, très rare dans la dermatite atopique et peu fréquent dans la pelade. Dans les études cliniques portant sur la dermatite atopique, le nombre d’infections cutanées nécessitant un traitement antibiotique était inférieur avec le baricitinib comparativement au placebo.
L’incidence d’infections graves avec le baricitinib a été similaire à celle du placebo. L’incidence des infections graves est restée stable durant l’exposition à long terme. Le taux d’incidence global d’infections graves dans le programme d’études cliniques du baricitinib a été de 3,2 pour 100 patients-années dans la polyarthrite rhumatoïde, de 2,1 pour 100 patients-années dans la dermatite atopique et de 0,8 pour 100 patients-années dans la pelade. Des cas de pneumonies graves et de zonas graves sont survenus de façon peu fréquente chez des patients atteints de polyarthrite rhumatoïde.
Élévations des transaminases hépatiques
Des augmentations dose-dépendantes de l’activité de l’ALAT et de l’ASAT sanguines ont été rapportées dans les études étendues à 16 semaines. Les élévations d’ALAT/ASAT moyennes sont restées stables dans le temps. La plupart des cas d’élévations des transaminases hépatiques ≥ 3 x LSN étaient asymptomatiques et transitoires.
Chez des patients atteints de polyarthrite rhumatoïde, l’association du baricitinib avec des médicaments potentiellement hépatotoxiques, comme le méthotrexate, a entraîné une fréquence accrue de ces élévations.
Élévations des lipides
Dans les données intégrant les études cliniques portant sur la polyarthrite rhumatoïde, la dermatite atopique et la pelade, le traitement par baricitinib a été associé à des augmentations dose-dépendantes des paramètres lipidiques, incluant le cholestérol total, le LDL-cholestérol et le cholestérol des lipoprotéines de haute densité (HDL-cholestérol). Aucun changement du rapport LDL/HDL n’a été observé. Les élévations ont été observées à 12 semaines et sont restées stables par la suite avec une valeur plus élevée que celle observée à l’inclusion, y compris pendant l’étude d’extension à long terme dans la polyarthrite rhumatoïde. Le cholestérol total et le LDL-cholestérol moyens ont augmenté jusqu’à la 52ème semaine chez les patients atteints de dermatite atopique et de pelade. Dans les études cliniques portant sur la polyarthrite rhumatoïde, le traitement par baricitinib a été associé à des augmentations doses-dépendantes des triglycérides. Il n’y a pas eu d’augmentation des triglycérides dans les études cliniques portant sur la dermatite atopique et la pelade.
Les élévations du LDL-cholestérol ont été réversibles sous statines pour revenir aux taux avant mise sous traitement.
Créatine phosphokinase (CPK)
Le traitement par baricitinib a été associé à des augmentations doses-dépendantes des CPK. La CPK moyenne a augmenté à la semaine 4 et est restée à une valeur plus élevée que celle observée à l’inclusion. Dans toutes les indications, la plupart des augmentations de CPK > 5 x LSN ont été transitoires et n’ont pas nécessité l’arrêt du traitement.
Dans les études cliniques, aucun cas de rhabdomyolyse n’a été confirmé.
Neutropénie
Le nombre moyen de polynucléaires neutrophiles a diminué à 4 semaines puis est resté stable dans le temps à une valeur plus basse qu’à l’inclusion. Aucune corrélation claire n’a été établie entre les neutropénies et la survenue d’infections graves. En revanche, dans les études cliniques, le traitement a été interrompu en cas de découverte d’un nombre absolu de polynucléaires neutrophiles < 1 x 109 cellules/L.
Thrombocytose
Des augmentations doses-dépendantes du nombre moyen de plaquettes ont été observées et sont restées stables dans le temps à une valeur plus élevée que celle observée à l’inclusion.
Population pédiatrique
Arthrite juvénile idiopathique
Au total, 220 patients âgés de 2 ans à moins de 18 ans ont été exposés à une dose de baricitinib dans le cadre du programme d'essais cliniques dans l'arthrite juvénile idiopathique, ce qui représente 326 patients-années d'exposition.
Chez les patients pédiatriques traités par le baricitinib au cours de la période de retrait de l’étude randomisée, en double aveugle, contrôlée versus placebo dans l'arthrite juvénile idiopathique (n = 82), les céphalées ont été très fréquentes (11 %), les neutropénies < 1 000 cellules/mm3 ont été fréquentes (2,4 %, un patient) et les embolies pulmonaires ont été fréquentes (1,2 %, un patient).
Dermatite atopique pédiatrique
L’évaluation de la sécurité chez les enfants et adolescents est basée sur les données de tolérance issues de l’étude clinique de phase III BREEZE-AD-PEDS dans laquelle 466 patients âgés entre 2 et 18 ans ont reçu une dose de baricitinib. Globalement, le profil de tolérance chez ces patients était comparable à celui observé dans la population adulte. La neutropénie (< 1 x 109 cellules/L) était plus fréquente (1,7 %) que chez les adultes.
Pelade (Alopecia areata) chez l’adolescent
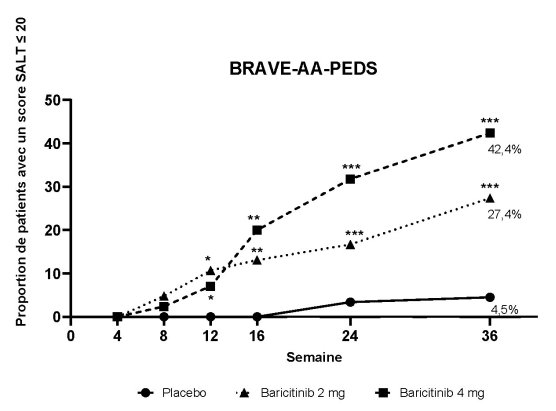
Au total, 245 patients âgés de 12 à moins de 18 ans ont été exposés à une dose de baricitinib dans la phase III de l’étude BRAVE-AA-PEDS. Parmi eux, 85 patients ont reçu la dose de 4 mg durant la période contrôlée par placebo. Globalement, le profil de tolérance chez ces patients était comparable à celui observé dans la population adulte. L’acné (10,6 %) et la neutropénie (3,6 %) (< 1 x 109 cellules/L) étaient plus fréquentes que chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique
Agence fédérale des médicaments et des produits de santé, www.afmps.be, Division Vigilance: Site internet: www.notifieruneffetindesirable.be, e-mail: adr@fagg-afmps.be.
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet : www.guichet.lu/pharmacovigilance.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Eli Lilly Nederland B.V., Orteliuslaan 1000, 3528 BD Utrecht, Pays-Bas.
8. NUMÉROS D’AUTORISATION DE MISE SUR LE MARCHÉ
Olumiant 1 mg comprimés pelliculés
EU/1/16/1170/017
EU/1/16/1170/018
EU/1/16/1170/019
Olumiant 2 mg comprimés pelliculés
EU/1/16/1170/001
EU/1/16/1170/002
EU/1/16/1170/003
EU/1/16/1170/004
EU/1/16/1170/005
EU/1/16/1170/006
EU/1/16/1170/007
EU/1/16/1170/008
Olumiant 4 mg comprimés pelliculés
EU/1/16/1170/009
EU/1/16/1170/010
EU/1/16/1170/011
EU/1/16/1170/012
EU/1/16/1170/013
EU/1/16/1170/014
EU/1/16/1170/015
EU/1/16/1170/016
10. DATE DE MISE À JOUR DU TEXTE : 27 AVRIL 2026
STATUT LEGAL DE DELIVRANCE Médicament sur prescription médicale restreinte.
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/.
1
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3593324 | OLUMIANT 2MG COMP PELL 28 X 2MG | L04AA37 | € 910,3 | - | Oui | € 12,8 | € 8,5 |
| 3593332 | OLUMIANT 2MG COMP PELL 84 X 2MG | L04AA37 | € 2402,8 | - | Oui | € 15,9 | € 10,5 |
| 3593340 | OLUMIANT 4MG COMP PELL 28 X 4MG | L04AA37 | € 910,3 | - | Oui | € 12,8 | € 8,5 |
| 3593357 | OLUMIANT 4MG COMP PELL 84 X 4MG | L04AA37 | € 2402,8 | - | Oui | € 15,9 | € 10,5 |