1. DÉNOMINATION DU MÉDICAMENT
XELJANZ 5 mg, comprimés pelliculés
XELJANZ 10 mg, comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
XELJANZ 5 mg, comprimés pelliculés
Chaque comprimé pelliculé contient du citrate de tofacitinib, équivalant à 5 mg de tofacitinib.
Excipient à effet notoire
Chaque comprimé pelliculé contient 59,44 mg de lactose.
XELJANZ 10 mg, comprimés pelliculés
Chaque comprimé pelliculé contient du citrate de tofacitinib, équivalant à 10 mg de tofacitinib.
Excipient à effet notoire
Chaque comprimé pelliculé contient 118,88 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé)
XELJANZ 5 mg, comprimés pelliculés
Comprimé blanc, rond, de 7,9 mm de diamètre, portant l’inscription « Pfizer » sur une face et « JKI 5 » sur l’autre face.
XELJANZ 10 mg, comprimés pelliculés
Comprimé bleu, rond, de 9,5 mm de diamètre, portant l’inscription « Pfizer » sur une face et « JKI 10 » sur l’autre face.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Polyarthrite rhumatoïde
Tofacitinib en association au méthotrexate (MTX) est indiqué dans le traitement de la polyarthrite rhumatoïde (PR) active, modérée à sévère chez les patients adultes ayant présenté une réponse inadéquate ou une intolérance à un ou plusieurs traitements de fond antirhumatismaux (DMARDs : Disease‑Modifying Antirheumatic Drugs) (voir rubrique 5.1). Tofacitinib peut être administré en monothérapie en cas d’intolérance au MTX ou lorsque le traitement avec le MTX est inadapté (voir rubriques 4.4 et 4.5).
Rhumatisme psoriasique
Tofacitinib en association au MTX est indiqué dans le traitement du rhumatisme psoriasique (RP) actif chez les patients adultes ayant présenté une réponse inadéquate ou une intolérance à un traitement de fond antirhumatismal (DMARD) antérieur (voir rubrique 5.1).
Spondylarthrite ankylosante
Tofacitinib est indiqué dans le traitement des patients adultes atteints de spondylarthrite ankylosante (SA) active qui n’ont pas répondu de manière adéquate au traitement conventionnel.
Rectocolite hémorragique
Tofacitinib est indiqué dans le traitement de la rectocolite hémorragique (RCH) active modérée à sévère chez les patients adultes ayant présenté une réponse inadéquate, une perte de réponse ou une intolérance soit au traitement conventionnel, soit à un agent biologique (voir rubrique 5.1).
Arthrite juvénile idiopathique (AJI)
Tofacitinib est indiqué dans le traitement de l’arthrite juvénile idiopathique polyarticulaire active (polyarthrite à facteur rhumatoïde positif [RF+] ou négatif [RF-] et oligoarthrite étendue) et du rhumatisme psoriasique (RP) juvénile chez les patients âgés de 2 ans et plus, ayant présenté une réponse inadéquate à un traitement par DMARD antérieur.
Tofacitinib peut être administré en association au méthotrexate (MTX) ou en monothérapie en cas d’intolérance au MTX ou lorsque la poursuite du traitement avec le MTX est inadaptée.
4.2 Posologie et mode d’administration
Le traitement doit être initié et surveillé par un médecin spécialiste ayant l’expérience du diagnostic et du traitement des affections pour lesquelles tofacitinib est indiqué.
Posologie
Polyarthrite rhumatoïde et rhumatisme psoriasique
La dose recommandée est de 5 mg en comprimés pelliculés administrée deux fois par jour, à ne pas dépasser.
Aucun ajustement posologique n’est nécessaire lors d’une utilisation en association avec le MTX.
Pour obtenir des informations sur le relai entre les comprimés pelliculés de tofacitinib et les comprimés à libération prolongée de tofacitinib, voir le tableau 1.
Tableau 1 : Relai entre les comprimés pelliculés de tofacitinib et les comprimés à libération prolongée de tofacitinib
Relai entre le tofacitinib 5 mg, comprimés pelliculés et le tofacitinib 11 mg, comprimés à libération prolongéea | Le traitement par tofacitinib 5 mg, comprimés pelliculés, deux fois par jour, et par tofacitinib 11 mg, comprimés à libération prolongée, une fois par jour, peut être interverti le jour suivant l’administration de la dernière dose de l’un ou l’autre des comprimés. |
a Voir rubrique 5.2 pour la comparaison de la pharmacocinétique des formulations à libération prolongée et des formulations pelliculées. | |
Spondylarthrite ankylosante
La dose recommandée de tofacitinib est de 5 mg administrée deux fois par jour.
Rectocolite hémorragique
Traitement d’induction
La dose recommandée est de 10 mg administrée par voie orale deux fois par jour pour le traitement d’induction pendant 8 semaines.
Pour les patients n’ayant pas obtenu un bénéfice thérapeutique adéquat à la semaine 8, la dose d’induction de 10 mg administrée deux fois par jour peut être prolongée pendant 8 semaines supplémentaires (16 semaines au total) suivie par une dose de 5 mg administrée deux fois par jour pour le traitement d’entretien. Le traitement d’induction avec tofacitinib doit être interrompu chez tout patient ne montrant aucun signe de bénéfice thérapeutique à la semaine 16.
Traitement d’entretien
La dose recommandée pour le traitement d’entretien est de 5 mg de tofacitinib administrée par voie orale deux fois par jour.
Le tofacitinib 10 mg deux fois par jour pour le traitement d’entretien n’est pas recommandé chez les patients atteints de RCH présentant des facteurs de risque connus de maladie thromboembolique veineuse (MTEV), d’événements cardiovasculaires indésirables majeurs (MACE) et de tumeur maligne, sauf en l’absence d’alternative thérapeutique appropriée (voir rubriques 4.4 et 4.8).
Chez les patients atteints de RCH qui ne sont pas à risque élevé de MTEV, de MACE et de tumeur maligne (voir rubrique 4.4), une administration de 10 mg de tofacitinib par voie orale deux fois par jour peut être envisagée si le patient présente une diminution de la réponse au tofacitinib 5 mg deux fois par jour et s’il n’a pas répondu aux autres options thérapeutiques pour la rectocolite hémorragique (RCH), telles qu’un traitement par un inhibiteur du facteur de nécrose tumorale (inhibiteur du TNF). Le tofacitinib 10 mg deux fois par jour pour le traitement d’entretien doit être utilisé pendant la plus courte durée possible. La dose efficace la plus faible permettant le maintien de la réponse doit être utilisée.
Chez les patients qui ont répondu au traitement avec tofacitinib, les corticostéroïdes peuvent être réduits et/ou interrompus conformément au cadre des soins habituels.
Reprise du traitement en cas de RCH
Si le traitement a été interrompu, la reprise d’un traitement avec tofacitinib peut être envisagée. En cas de perte de réponse, une réinduction avec tofacitinib 10 mg administré deux fois par jour peut être envisagée. Au cours des études cliniques, la durée de la période d’interruption du traitement a pu atteindre 1 an. L’efficacité peut être récupérée par un traitement de 8 semaines à la dose de 10 mg administrée deux fois par jour (voir rubrique 5.1).
AJI polyarticulaire et RP juvénile (enfants âgés de 2 à 18 ans)
Tofacitinib peut être utilisé en monothérapie ou en association avec le MTX.
La dose recommandée chez les patients âgés de 2 ans et plus est basée sur les catégories de poids suivantes :
Tableau 2 : Dose de tofacitinib pour les patients atteints d’arthrite juvénile idiopathique polyarticulaire et de RP juvénile âgés de deux ans et plus
Poids corporel (kg) | Schéma posologique |
10 ‑ < 20 | 3,2 mg (3,2 mL de solution buvable) deux fois par jour |
20 ‑ < 40 | 4 mg (4 mL de solution buvable) deux fois par jour |
≥ 40 | 5 mg (5 mL de solution buvable ou comprimé pelliculé de 5 mg) deux fois par jour |
Les patients pesant ≥ 40 kg traités par la solution buvable de 5 mL de tofacitinib deux fois par jour peuvent passer au tofacitinib, 5 mg comprimés pelliculés deux fois par jour. Les patients pesant < 40 kg ne peuvent pas passer à une autre formulation que la solution buvable de tofacitinib.
Interruption et arrêt du traitement chez les patients adultes et pédiatriques
Le traitement avec tofacitinib doit être interrompu si un patient développe une infection grave jusqu’à ce que cette dernière soit contrôlée.
L’interruption du traitement peut être nécessaire afin de contrôler les anomalies biologiques dose-dépendantes, incluant la lymphopénie, la neutropénie, et l’anémie. Comme décrit dans les Tableaux 3, 4 et 5 ci-dessous, les recommandations d’interruption temporaire ou d’arrêt définitif du traitement sont déterminées selon la sévérité des anomalies biologiques (voir rubrique 4.4).
Il est recommandé de ne pas initier le traitement chez les patients présentant une numération absolue des lymphocytes (NAL) inférieure à 750 cellules/mm3.
Tableau 3 : Faible numération absolue des lymphocytes
Faible numération absolue des lymphocytes (NAL) (voir rubrique 4.4) | |
Valeur biologique | Recommandation |
NAL supérieure ou égale à 750 | Le traitement doit être maintenu. |
NAL 500-750 | Pour une réduction persistante dans cette fourchette (2 valeurs séquentielles dans cette fourchette au cours des tests de routine), le traitement doit être réduit ou interrompu. |
NAL inférieure à 500 | Si cette valeur est confirmée par un nouveau test dans les 7 jours qui suivent, le traitement doit être arrêté. |
Il est recommandé de ne pas initier le traitement chez les patients adultes présentant une numération absolue des neutrophiles (NAN) inférieure à 1 000 cellules/mm3. Il est recommandé de ne pas initier le traitement chez les patients pédiatriques présentant une numération absolue des neutrophiles (NAN) inférieure à 1 200 cellules/mm3.
Tableau 4 : Faible numération absolue des neutrophiles
Faible numération absolue des neutrophiles (NAN) (voir rubrique 4.4) | |
Valeur biologique | Recommandation |
NAN supérieure à 1 000 | Le traitement doit être maintenu. |
NAN 500 – 1 000 | Pour les réductions persistantes dans cette fourchette (2 valeurs séquentielles dans cette fourchette au cours des tests de routine), le traitement doit être réduit ou interrompu. |
NAN inférieure à 500 | Si cette valeur biologique est confirmée par un nouveau test dans les 7 jours qui suivent, le traitement doit être arrêté. |
Il est recommandé de ne pas initier le traitement chez les patients adultes présentant un taux d’hémoglobine inférieur à 9 g/dL. Il est recommandé de ne pas initier le traitement chez les patients pédiatriques présentant un taux d’hémoglobine inférieur à 10 g/dL.
Tableau 5 : Faible taux d’hémoglobine
Faible taux d’hémoglobine (voir rubrique 4.4) | |
Valeur biologique | Recommandation |
Diminution inférieure ou égale à 2 g/dL et taux supérieur ou égal à 9,0 g/dL | Le traitement doit être maintenu. |
Diminution supérieure à 2 g/dL ou taux inférieur à 8,0 g/dL | Le traitement doit être interrompu jusqu’à ce que les valeurs de l’hémoglobine se soient normalisées. |
Interactions
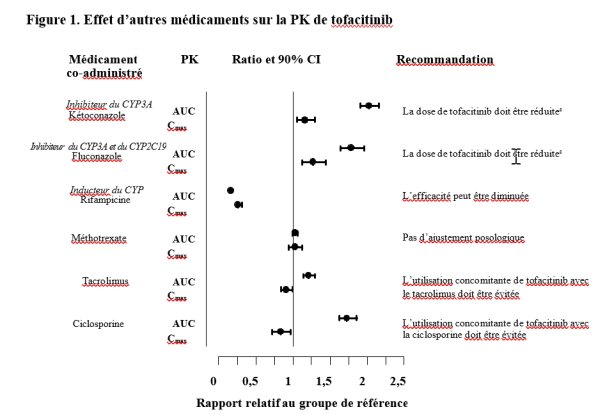
La dose quotidienne totale de tofacitinib doit être réduite de moitié chez les patients recevant des inhibiteurs puissants du cytochrome (CYP) P450 3A4 (par ex., le kétoconazole) et chez les patients recevant un ou plusieurs médicaments concomitants entraînant une inhibition modérée du CYP3A4 ainsi qu’une inhibition puissante du CYP2C19 (par ex., le fluconazole) (voir rubrique 4.5) comme suit :
- La dose de tofacitinib doit être réduite à 5 mg une fois par jour chez les patients recevant 5 mg deux fois par jour (patients adultes et pédiatriques).
- La dose de tofacitinib doit être réduite à 5 mg deux fois par jour chez les patients recevant 10 mg deux fois par jour (patients adultes).
Seulement pour les patients pédiatriques :
Les données disponibles suggèrent qu’une amélioration clinique est observée dans les 18 semaines suivant l’initiation du traitement par tofacitinib. La poursuite du traitement doit être reconsidérée avec précaution chez un patient ne présentant aucune amélioration clinique dans ce délai.
Interruption du traitement dans la SA
Les données disponibles suggèrent qu’une amélioration clinique de la SA est observée dans les 16 semaines suivant l’instauration du traitement par tofacitinib. La poursuite du traitement doit être reconsidérée avec précaution chez un patient ne présentant aucune amélioration clinique dans ce délai.
Populations particulières
Personnes âgées
Aucun ajustement posologique n’est nécessaire chez les patients âgés de 65 ans et plus. Les données disponibles chez les patients âgés de 75 ans et plus sont limitées. Voir rubrique 4.4 pour l’utilisation chez les patients âgés de 65 ans et plus.
Insuffisance hépatique
Tableau 6 : Ajustement posologique en cas d’insuffisance hépatique
Catégorie d’insuffisance hépatique | Classification | Ajustement posologique en cas d’insuffisance hépatique pour des comprimés de dosage différent |
Légère | Classe A de Child-Pugh | Aucun ajustement posologique n’est nécessaire. |
Modérée | Classe B de Child-Pugh | La dose doit être réduite à 5 mg une fois par jour quand la dose indiquée en présence d’une fonction hépatique normale est de 5 mg deux fois par jour. |
Sévère | Classe C de Child-Pugh | Tofacitinib ne doit pas être utilisé chez les patients présentant une insuffisance hépatique sévère (voir rubrique 4.3). |
Insuffisance rénale
Tableau 7 : Ajustement posologique en cas d’insuffisance rénale
Catégorie d’insuffisance rénale | Clairance de la créatinine | Ajustement posologique en cas d’insuffisance rénale pour des comprimés de dosage différent |
Légère | 50-80 mL/min | Aucun ajustement posologique n’est nécessaire. |
Modérée | 30-49 mL/min | Aucun ajustement posologique n’est nécessaire. |
Sévère (y compris patients hémodialysés) | < 30 mL/min | La dose doit être réduite à 5 mg une fois par jour quand la dose indiquée en présence d’une fonction rénale normale est de 5 mg deux fois par jour. |
Population pédiatrique
La sécurité et l’efficacité du tofacitinib chez les enfants âgés de moins de 2 ans atteints d’AJI polyarticulaire et de RP juvénile n’ont pas été établies.Aucune donnée n’est disponible.
La sécurité et l’efficacité du tofacitinib chez les enfants âgés de moins de 18 ans atteints d’autres indications (par exemple, rectocolite hémorragique) n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie orale.
Tofacitinib est administré par voie orale, avec ou sans nourriture.
Pour les patients ayant des difficultés à avaler, les comprimés de tofacitinib peuvent être écrasés et pris avec de l'eau.
4.3 Contre-indications
- Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
- Tuberculose (TB) active, infections graves telles qu’une septicémie ou des infections opportunistes (voir rubrique 4.4).
- Insuffisance hépatique sévère (voir rubrique 4.2).
- Grossesse et allaitement (voir rubrique 4.6).
4.8 Effets indésirables
Résumé du profil de tolérance
Polyarthrite rhumatoïde
Les effets indésirables graves les plus fréquents étaient des infections graves (voir rubrique 4.4).
Dans l’étude de tolérance à long terme sur l’ensemble de la population exposée, les infections graves les plus fréquemment rapportées au cours du traitement avec tofacitinib étaient les suivantes : pneumonie (1,7 %), zona (0,6 %), infections des voies urinaires (0,4 %), cellulite (0,4 %), diverticulite (0,3 %) et appendicite (0,2 %). Les infections opportunistes suivantes ont été rapportées chez des patients traités avec tofacitinib : TB et autres infections mycobactériennes, cryptococcose, histoplasmose, candidose œsophagienne, zona multimétamérique, infection à cytomégalovirus, infections au virus BK et listériose. Chez certains patients, l’infection se présentait sous forme disséminée plutôt que localisée. D’autres infections graves qui n’ont pas été rapportées au cours des études cliniques pourraient également survenir (par ex., coccidioïdomycose).
Au cours des études cliniques en double aveugle contrôlées contre placebo ou MTX, les évènements indésirables les plus fréquemment rapportés au cours des 3 premiers mois étaient les suivants : céphalées (3,9 %), infections des voies respiratoires supérieures (3,8 %), infection virale des voies aériennes supérieures (3,3 %), diarrhée (2,9 %), nausées (2,7 %) et hypertension (2,2 %).
Dans les essais en double aveugle contrôlés contre placebo ou MTX, la proportion de patients arrêtant le traitement en raison d’un évènement indésirable lors des 3 premiers mois était de 3,8 % pour les patients sous tofacitinib. Les infections les plus fréquentes, entraînant une interruption du traitement au cours des 3 premiers mois dans les études cliniques contrôlées, étaient le zona (0,19 %) et la pneumonie (0,15 %).
Rhumatisme psoriasique
Globalement, le profil de tolérance observé chez les patients atteints de RP actif traités avec tofacitinib était conforme au profil de tolérance observé chez les patients atteints de PR traités avec tofacitinib.
Spondylarthrite ankylosante
Globalement, le profil de tolérance observé chez les patients atteints de SA active traités avec tofacitinib était conforme au profil de tolérance observé chez les patients atteints de PR traités avec tofacitinib.
Rectocolite hémorragique
Les effets indésirables les plus fréquemment rapportés chez les patients recevant tofacitinib 10 mg deux fois par jour au cours des études d’induction étaient des céphalées, des rhinopharyngites, des nausées et des arthralgies.
Au cours des études d’induction et d’entretien, dans les groupes de traitement tofacitinib et placebo, les catégories les plus fréquentes d’effets indésirables graves étaient les affections gastro-intestinales et les infections, et l’effet indésirable grave le plus fréquent était l’aggravation de la RCH.
Globalement, le profil de sécurité observé chez les patients atteints de RCH traités avec tofacitinib était cohérent avec le profil de sécurité de tofacitinib dans l’indication de PR.
Liste tabulée des effets indésirables
Les effets indésirables listés dans le tableau ci-dessous proviennent des études cliniques menées chez des patients atteints de PR, de RP, de SA et de RCH et sont présentés par classe de systèmes d’organes (SOC) et catégories de fréquence, définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), ou indéterminée (impossible à estimer sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 8 : Effets indésirables
Classe de système d'organes | Fréquent | Peu fréquent | Rare | Très rare | Indéterminée (impossible à estimer sur la base des données disponibles) |
Infections et infestations | Pneumonie | Tuberculose | Septicémie | Tuberculose du système nerveux central |
|
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) |
| Cancer du poumon | Lymphome |
|
|
Affections hématologiques et du système lymphatique | Lymphopénie | Leucopénie |
|
|
|
Affections du système immunitaire |
|
|
|
| Hypersensibilité* |
Troubles du métabolisme et de la nutrition |
| Dyslipidémie |
|
|
|
Affections psychiatriques |
| Insomnie |
|
|
|
Affections du système nerveux | Céphalées | Paresthésies |
|
|
|
Affections cardiaques |
| Infarctus du myocarde |
|
|
|
Affections vasculaires | Hypertension | Maladie thromboembolique veineuse** |
|
|
|
Affections respiratoires, thoraciques et médiastinales | Toux | Dyspnée |
|
|
|
Affections gastro-intestinales | Douleurs abdominales |
|
|
|
|
Affections hépatobiliaires |
| Stéatose hépatique | Exploration fonctionnelle hépatique anormale |
|
|
Affections de la peau et du tissu sous-cutané | Éruption cutanée Acné | Érythème |
|
|
|
Affections musculo-squelettiques et systémiques | Arthralgie | Tuméfaction articulaire | Douleur musculo-squelettique |
|
|
Troubles généraux et anomalies au site d’administration | Œdème périphérique | Fièvre |
|
|
|
Investigations | Créatine phosphokinase sanguine augmentée | Créatinine sanguine augmentée |
|
|
|
Lésions, intoxications et complications liées aux interventions |
| Entorse d’un ligament |
|
|
|
* Données issues des notifications spontanées
** La maladie thromboembolique veineuse comprend l’embolie pulmonaire (EP), la thrombose veineuse profonde (TVP), la thrombose veineuse rétinienne, et la Thrombose des Sinus Veineux Cérébraux.
Description de certains effets indésirables
Maladie thromboembolique veineuse
Polyarthrite rhumatoïde
Au cours d’une vaste étude randomisée post-autorisation (N = 4 362) évaluant la sécurité chez des patients atteints de polyarthrite rhumatoïde âgés de 50 ans et plus et présentant au moins un facteur de risque cardiovasculaire (CV) supplémentaire, une incidence accrue et dose-dépendante de MTEV a été observée chez les patients traités par tofacitinib, comparativement aux inhibiteurs du TNF (voir rubrique 5.1). La majorité de ces événements ont été graves et certains ont entraîné des décès. Les taux d’incidence (IC à 95 %) des EP pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF étaient respectivement de 0,17 (0,08 – 0,33), de 0,50 (0,32 – 0,74) et de 0,06 (0,01 – 0,17) patient avec des événements pour 100 patient‑années. Comparativement aux inhibiteurs du TNF, le hazard ratio (HR) pour l’EP a été respectivement de 2,93 (0,79 – 10,83) et de 8,26 (2,49 – 27,43) pour le tofacitinib 5 mg deux fois par jour et le tofacitinib 10 mg deux fois par jour (voir rubrique 5.1). Chez les patients traités par tofacitinib chez lesquels une EP a été observée, la majorité (97 %) présentait des facteurs de risque de MTEV.
Spondylarthrite ankylosante
Au cours des études cliniques contrôlées randomisées combinées de phase 2 et de phase 3, aucun événement de MTEV n’a été observé chez les 420 patients (233 patient-années d’observation) recevant du tofacitinib jusqu’à 48 semaines.
Rectocolite hémorragique (RCH)
Au cours de l’essai d’extension en cours portant sur la RCH, des cas d’EP et de TVP ont été observés chez des patients utilisant 10 mg de tofacitinib deux fois par jour et présentant un ou plusieurs facteurs de risque de MTEV sous-jacents.
Infections
Polyarthrite rhumatoïde
Au cours des études cliniques contrôlées de phase 3, les taux d’infections sur 0 – 3 mois dans les groupes recevant tofacitinib en monothérapie 5 mg deux fois par jour (616 patients au total) et 10 mg deux fois par jour (642 patients au total) étaient de 16,2 % (100 patients) et 17,9 % (115 patients), respectivement, contre 18,9 % (23 patients) dans le groupe placebo (122 patients au total). Au cours des études cliniques contrôlées de phase 3 menées chez des patients recevant un traitement de fond concomitant par DMARD, les taux d’infections sur 0 – 3 mois dans les groupes tofacitinib 5 mg deux fois par jour plus DMARD (973 patients au total) et tofacitinib 10 mg deux fois par jour plus DMARD (969 patients au total) étaient de 21,3 % (207 patients) et 21,8 % (211 patients), respectivement, contre 18,4 % (103 patients) dans le groupe placebo plus DMARD (559 patients au total).
Les infections les plus fréquemment rapportées étaient les infections des voies respiratoires supérieures et les rhinopharyngites (3,7 % et 3,2 %, respectivement).
Le taux d’incidence globale des infections sous tofacitinib dans la population d’étude de tolérance à long terme sur l’ensemble de la population exposée (au total 4 867 patients) était de 46,1 patients avec des événements pour 100 patient-années (43,8 et 47,2 patients avec des événements pour les groupes 5 mg et 10 mg deux fois par jour, respectivement). Pour les patients en monothérapie (1 750 patients au total), les taux étaient de 48,9 et 41,9 patients avec des événements pour 100 patient-années pour les groupes 5 mg et 10 mg deux fois par jour, respectivement. Pour les patients recevant un traitement de fond concomitant par DMARD (3 117 patients au total), les taux étaient de 41,0 et 50,3 patients avec des événements pour 100 patient-années pour les groupes 5 mg et 10 mg deux fois par jour, respectivement.
Spondylarthrite ankylosante
Au cours des études cliniques combinées de phase 2 et de phase 3, pendant la période contrôlée contre placebo allant jusqu’à 16 semaines, la fréquence des infections dans le groupe tofacitinib 5 mg deux fois par jour (185 patients) était de 27,6 % et la fréquence dans le groupe placebo (187 patients) était de 23,0 %. Au cours des études cliniques combinées de phase 2 et de phase 3, parmi les 316 patients traités par tofacitinib 5 mg deux fois par jour pendant une période allant jusqu’à 48 semaines, la fréquence des infections était de 35,1 %.
Rectocolite hémorragique
Au cours des études d’induction randomisées de phase 2/3, de 8 semaines, les taux de patients présentant des infections étaient de 21,1 % (198 patients) dans le groupe tofacitinib 10 mg deux fois par jour contre 15,2 % (43 patients) dans le groupe placebo. Dans l’étude d’entretien randomisée de phase 3, de 52 semaines, les taux de patients présentant des infections étaient de 35,9 % (71 patients) dans le groupe tofacitinib 5 mg deux fois par jour et de 39,8 % (78 patients) dans le groupe tofacitinib 10 mg deux fois par jour, contre 24,2 % (48 patients) dans le groupe placebo.
Sur l’ensemble de l’expérience thérapeutique disponible avec tofacitinib, les infections les plus fréquemment rapportées étaient les rhinopharyngites, qui sont survenues chez 18,2 % des patients (211 patients).
Sur l’ensemble de l’expérience thérapeutique disponible avec tofacitinib, le taux d’incidence global des infections était de 60,3 événements pour 100 patient-années (concernant 49,4 % des patients ; pour un total de 572 patients).
Infections graves
Polyarthrite rhumatoïde
Au cours des études cliniques contrôlées de 6 mois et de 24 mois, le taux d’infections graves dans le groupe tofacitinib 5 mg deux fois par jour en monothérapie était de 1,7 patient avec des événements pour 100 patient-années. Dans le groupe tofacitinib 10 mg deux fois par jour en monothérapie, le taux était de 1,6 patient avec des événements pour 100 patient‑années, le taux était de 0 événement pour 100 patient-années pour le groupe placebo, et le taux était de 1,9 patient avec des événements pour 100 patient-années pour le groupe MTX.
Au cours des études de 6 mois, 12 mois et 24 mois, les taux d’infections graves dans les groupes tofacitinib 5 mg deux fois par jour plus DMARD et tofacitinib 10 mg deux fois par jour plus DMARD étaient de 3,6 et 3,4 patients avec des événements pour 100 patient-années, respectivement, contre 1,7 patient avec des événements pour 100 patient-années dans le groupe placebo plus DMARD.
Dans la population d’étude de tolérance à long terme sur l’ensemble de la population exposée, les taux globaux d’infections graves étaient de 2,4 et 3,0 patients avec des événements pour 100 patient-années pour les groupes tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Les infections graves les plus fréquentes comprenaient la pneumonie, le zona, l’infection des voies urinaires, la cellulite, la gastro-entérite et la diverticulite. Des cas d’infections opportunistes ont été rapportés (voir rubrique 4.4).
Au cours d’une vaste étude de sécurité randomisée post-autorisation (N = 4 362) menée chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, une augmentation dose-dépendante des infections graves a été observée avec le tofacitinib par rapport aux inhibiteurs du TNF (voir rubrique 4.4).
Les taux d’incidence (IC à 95 %) des infections graves pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF étaient de 2,86 (2,41 ; 3,37), de 3,64 (3,11 ; 4,23) et de 2,44 (2,02 ; 2,92) patients avec des événements pour 100 patient-années, respectivement. Par rapport aux inhibiteurs du TNF, le hazard ratio (HR) pour les infections graves était de 1,17 (0,92 ; 1,50) et de 1,48 (1,17 ; 1,87) pour le tofacitinib 10 mg deux fois par jour et le tofacitinib 5 mg deux fois par jour, respectivement.
Spondylarthrite ankylosante
Au cours des études cliniques combinées de phase 2 et de phase 3, parmi les 316 patients traités avec le tofacitinib 5 mg deux fois par jour pendant une durée maximale de 48 semaines, une infection grave (méningite aseptique) a été observée, ce qui représente un taux de 0,43 patient présentant des événements pour 100 patient-années.
Rectocolite hémorragique
Les taux d’incidence et les types d’infections graves au cours des études cliniques de la RCH ont été généralement similaires à ceux qui avaient été rapportés au cours des études cliniques de la PR pour les groupes de traitement avec tofacitinib en monothérapie.
Infections graves chez les personnes âgées
Sur les 4 271 patients inclus dans les études I à VI sur la PR (voir rubrique 5.1), un total de 608 patients atteints de PR étaient âgés de 65 ans et plus, dont 85 patients âgés de 75 ans et plus. La fréquence des infections graves parmi les patients âgés de 65 ans et plus traités avec tofacitinib était supérieure à celle observée chez les patients âgés de moins de 65 ans (4,8 pour 100 patient-années versus 2,4 pour 100 patient-années, respectivement).
Au cours d’une vaste étude de sécurité randomisée post-autorisation (N = 4 362) menée chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, une augmentation des infections graves a été observée chez les patients âgés de 65 ans et plus pour le tofacitinib 10 mg deux fois par jour par rapport aux inhibiteurs du TNF et au tofacitinib 5 mg deux fois par jour (voir rubrique 4.4). Les taux d’incidence (IC à 95 %) des infections graves chez les patients âgés de ≥ 65 ans étaient de 4,03 (3,02 , 5,27), de 5,85 (4,64 , 7,30) et de 3,73 (2,81 , 4,85) patients avec des événements pour 100 patient-années pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF, respectivement.
Par rapport aux inhibiteurs du TNF, le hazard ratio (HR) pour les infections graves chez les patients âgés de ≥ 65 ans était de 1,08 (0,74 ; 1,58) et de 1,55 (1,10 ; 2,19) pour le tofacitinib 5 mg deux fois par jour et le tofacitinib 10 mg deux fois par jour, respectivement.
Infections graves au cours d’une étude de tolérance non interventionnelle post-autorisation
Les données d’une étude de tolérance non interventionnelle post-autorisation ayant évalué le tofacitinib chez des patients atteints de PR provenant d’un registre (US Corrona) ont révélé qu’un taux d’incidence numériquement plus élevé d’infections graves a été observé pour le comprimé à libération prolongée de 11 mg administré une fois par jour par rapport au comprimé pelliculé de 5 mg administré deux fois par jour. Les taux d’incidence bruts (IC à 95 %) (c’est-à-dire non ajustés en fonction de l’âge ou du sexe) depuis la disponibilité de chaque formulation à 12 mois après l’instauration du traitement ont été de 3,45 (1,93 ; 5,69) et de 2,78 (1,74 ; 4,21) et à 36 mois de 4,71 (3,08 ; 6,91) et de 2,79 (2,01 ; 3,77) patients avec des événements pour 100 patient-années dans le groupe des comprimés à libération prolongée de 11 mg administrés une fois par jour et des comprimés pelliculés de 5 mg administrés deux fois par jour, respectivement. Le rapport de risque non ajusté a été de 1,30 (IC à 95 % : 0,67 ; 2,50) à 12 mois et de 1,93 (IC à 95 % : 1,15 ; 3,24) à 36 mois pour la dose de 11 mg une fois par jour en comprimés à libération prolongée par rapport à la dose de 5 mg deux fois par jour en comprimés pelliculés. Les données reposent sur un petit nombre de patients avec des événements observés présentant des intervalles de confiance relativement larges et une durée de suivi limitée.
Réactivation virale
Les patients traités avec tofacitinib japonais ou coréens, les patients atteints de PR de longue date ayant précédemment reçu au moins deux DMARDs biologiques, les patients présentant une NAL inférieure à 1000 cellules/mm3, ou les patients traités par 10 mg deux fois par jour pourraient présenter un risque accru de zona (voir rubrique 4.4).
Au cours d’une vaste (N = 4 362) étude randomisée post-autorisation évaluant la sécurité chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, une augmentation des cas de zona a été observée chez les patients traités par tofacitinib par rapport aux inhibiteurs du TNF. Les taux d’incidence (IC à 95 %) du zona pour les traitements par tofacitinib 5 mg deux fois par jour, par tofacitinib 10 mg deux fois par jour et par les inhibiteurs du TNF étaient respectivement de 3,75 (3,22 ; 4,34), 3,94 (3,38 ; 4,57) et 1,18 (0,90 ; 1,52) patients avec événements pour 100 patient-années.
Analyses biologiques
Lymphocytes
Dans les études cliniques sur la PR contrôlées, des baisses confirmées de la NAL en dessous de 500 cellules/mm3 ont été rapportées chez 0,3 % des patients et des baisses de la NAL entre 500 et 750 cellules/mm3 chez 1,9% des patients pour toutes les doses confondues de tofacitinib 5 mg deux fois par jour et 10 mg deux fois par jour.
Dans la population d’étude de tolérance à long terme sur la PR, des baisses confirmées de la NAL en dessous de 500 cellules/mm3 ont été rapportées chez 1,3 % des patients et des baisses de la NAL entre 500 et 750 cellules/mm3 chez 8,4% des patients pour toutes les doses confondues de tofacitinib 5 mg deux fois par jour et 10 mg deux fois par jour.
Des taux confirmés de NAL inférieurs à 750 cellules/mm3 ont été associés à une incidence accrue d’infections graves (voir rubrique 4.4).
Dans les études cliniques de la RCH, les modifications de la NAL observées avec le traitement avec tofacitinib ont été similaires à celles qui avaient été observées au cours des études cliniques de la PR.
Neutrophiles
Dans les études cliniques sur la PR contrôlées, des baisses confirmées de la NAN en dessous de 1 000 cellules/mm3 ont été rapportées chez 0,08 % des patients pour toutes les doses confondues de tofacitinib 5 mg deux fois par jour et 10 mg deux fois par jour. Aucune baisse confirmée de la NAN en dessous de 500 cellules/mm3 n’a été observée parmi les groupes de traitement. Aucune relation claire n’a été établie entre la neutropénie et l’apparition d’infections graves.
Dans la population d’étude de tolérance à long terme sur la PR, le profil et l’incidence de baisses confirmées de la NAN sont restés cohérents avec ceux observés dans les études cliniques contrôlées (voir rubrique 4.4).
Dans les études cliniques de la RCH, les modifications de la NAN observées avec le traitement avec tofacitinib ont été similaires à celles qui avaient été observées au cours des études cliniques de la PR.
Plaquettes
Les patients des études cliniques contrôlées de phase 3 (PR, RP, SA, RCH) devaient avoir une numération plaquettaire ≥ 100 000 cellules/mm3 pour être éligibles au recrutement, par conséquent, aucune information n’est disponible pour les patients ayant une numération plaquettaire < 100 000 cellules/mm3 avant le début du traitement par tofacitinib.
Tests des enzymes hépatiques
Des hausses confirmées des enzymes hépatiques supérieures à 3 fois la limite supérieure de la normale (3 x LSN) ont été observées de façon peu fréquente chez les patients atteints de PR. Chez ces patients présentant une élévation des enzymes hépatiques, une modification du traitement, comme une diminution de la dose du DMARD concomitant, l’interruption de l’administration de tofacitinib ou la diminution de la dose de tofacitinib, a entraîné une baisse ou une normalisation des enzymes hépatiques.
Au cours de la période contrôlée de l’étude en monothérapie de phase 3 sur la PR (0 – 3 mois), (Étude I, voir rubrique 5.1), des élévations de l’ALAT supérieures à 3 x LSN ont été observées chez 1,65 %, 0,41 % et 0 % des patients prenant le placebo, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Dans cette étude, des élévations de l’ASAT supérieures à 3 x LSN ont été observées chez 1,65 %, 0,41 % et 0 % des patients recevant le placebo, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement.
Au cours de l’étude en monothérapie de phase 3 sur la PR (0 – 24 mois), (Étude VI, voir rubrique 5.1), des élévations de l’ALAT supérieures à 3 x LSN ont été observées chez 7,1 %, 3,0 % et 3,0 % des patients recevant le MTX, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Dans cette étude, des élévations de l’ASAT supérieures à 3 x LSN ont été observées chez 3,3 %, 1,6 % et 1,5 % des patients recevant le MTX, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement.
Au cours de la période contrôlée des études de phase 3 sur la PR chez des patients recevant un traitement de fond concomitant par DMARDs (0 – 3 mois), (Étude II–V, voir rubrique 5.1), des élévations de l’ALAT supérieures à 3 x LSN ont été observées chez 0,9 %, 1,24 % et 1,14 % des patients recevant le placebo, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Au cours de ces études, des élévations de l’ASAT supérieures à 3 x LSN ont été observées chez 0,72 %, 0,5 % et 0,31 % des patients recevant le placebo, tofacitinib 5 mg et 10 mg deux fois par jour, respectivement.
Au cours des études d’extension à long terme sur la PR, en monothérapie, des élévations de l’ALAT supérieures à 3 x LSN ont été observées chez 1,1 %, 1,4 % des patients recevant tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Des élévations de l’ASAT supérieures à 3 x LSN ont été observées chez <1.0% des patients dans chacun des deux groupes tofacitinib 5 mg et 10 mg deux fois par jour.
Au cours des études d’extension à long terme sur la PR, avec un traitement de fond concomitant par DMARD, des élévations de l’ALAT supérieures à 3 x LSN ont été observées chez 1,8 %, 1,6 % des patients recevant tofacitinib 5 mg et 10 mg deux fois par jour, respectivement. Des élévations de l’ASAT supérieures à 3 x LSN ont été observées chez <1.0% des patients dans chacun des deux groupes tofacitinib 5 mg et 10 mg deux fois par jour.
Au cours d’une vaste (N = 4 362) étude randomisée post-autorisation évaluant la sécurité chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, des élévations de l’ALAT supérieures ou égales à 3 x LSN ont été observées chez 6,01 %, 6,54 % et 3,77 % des patients traités respectivement par 5 mg de tofacitinib deux fois par jour, 10 mg de tofacitinib deux fois par jour et des inhibiteurs du TNF. Des élévations de l’ASAT supérieures ou égales à 3 x LSN ont été observées chez 3,21 %, 4,57 % et 2,38 % des patients traités respectivement par 5 mg de tofacitinib deux fois par jour, 10 mg de tofacitinib deux fois par jour et des inhibiteurs du TNF.
Au cours des études cliniques de la RCH, les modifications des tests des enzymes hépatiques observées avec le traitement avec tofacitinib ont été similaires à celles qui avaient été observées au cours des études cliniques de la PR.
Lipides
Des élévations des paramètres lipidiques (cholestérol total, LDL-cholestérol, HDL-cholestérol, triglycérides) ont d’abord été observées 1 mois après l’initiation du traitement avec tofacitinib au cours des études cliniques contrôlées en double aveugle portant sur la PR. Ces élévations ont été observées à un mois et sont restées stables par la suite.
Les variations des paramètres lipidiques observées entre l’inclusion et la fin de l’étude (6 – 24 mois) au cours des études cliniques contrôlées portant sur la PR, sont présentées ci-dessous :
- Le LDL-cholestérol moyen a augmenté de 15 % dans le bras tofacitinib 5 mg deux fois par jour et de 20 % dans le bras tofacitinib 10 mg deux fois par jour à 12 mois, et a augmenté de 16 % dans le bras tofacitinib 5 mg deux fois par jour et de 19 % dans le bras tofacitinib 10 mg deux fois par jour à 24 mois.
- Le HDL-cholestérol moyen a augmenté de 17 % dans le bras tofacitinib 5 mg deux fois par jour et de 18 % dans le bras tofacitinib 10 mg deux fois par jour à 12 mois, et a augmenté de 19 % dans le bras tofacitinib 5 mg deux fois par jour et de 20 % dans le bras tofacitinib 10 mg deux fois par jour à 24 mois.
À l’arrêt du traitement avec tofacitinib, les taux de lipides sont revenus aux valeurs initiales.
Les rapports LDL-cholestérol / HDL-cholestérol et les rapports apolipoprotéine B (ApoB)/ApoA1 moyens étaient globalement stables chez les patients traités avec tofacitinib.
Dans une étude clinique contrôlée sur la PR, les élévations du LDL-cholestérol et de l’ApoB sont revenues aux niveaux préthérapeutiques en réponse à un traitement par statines.
Dans les populations d’étude de tolérance à long terme sur la PR, les élévations des paramètres lipidiques sont restées cohérentes avec celles observées dans les essais cliniques contrôlés.
Au cours d’une vaste (N = 4 362) étude randomisée post-autorisation évaluant la sécurité chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, les modifications des paramètres lipidiques entre le début de l’étude et 24 mois après sont résumées ci-dessous :
- Le LDL-cholestérol moyen a augmenté de 13,80 %, 17,04 % et 5,50 % chez les patients traités par tofacitinib 5 mg deux fois par jour, par tofacitinib 10 mg deux fois par jour et par un inhibiteur du TNF, respectivement, au mois 12. Au mois 24, l’augmentation était respectivement de 12,71 %, 18,14 % et 3,64 %,
- Le HDL-cholestérol moyen a augmenté de 11,71 %, 13,63 % et 2,82 % chez les patients traités par tofacitinib 5 mg deux fois par jour, par tofacitinib 10 mg deux fois par jour et par inhibiteur du TNF, respectivement, au mois 12. Au mois 24, l’augmentation était respectivement de 11,58 %, 13,54 % et 1,42 %.
Au cours des études cliniques de la RCH, les modifications des lipides observées avec le traitement avec tofacitinib ont été similaires à celles qui avaient été observées au cours des études cliniques de la PR.
Infarctus du myocarde
Polyarthrite rhumatoïde
Au cours d’une vaste (N = 4 362) étude randomisée post-autorisation évaluant la sécurité chez des patients atteints de PR âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, les taux d’incidence (IC à 95 %) des infarctus du myocarde non fatals pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF étaient respectivement de 0,37 (0,22 – 0,57), de 0,33 (0,19 – 0,53) et de 0,16 (0,07 – 0,31) événements pour 100 patients-années. Quelques infarctus du myocarde fatals ont été rapportés avec des taux similaires chez les patients traités par tofacitinib comparativement aux inhibiteurs du TNF (voir rubriques 4.4 et 5.1). L’étude a nécessité le suivi d’au moins 1 500 patients pendant 3 ans.
Tumeurs malignes (sauf CCNM)
Polyarthrite rhumatoïde
Au cours d’une vaste (N = 4 362) étude randomisée post-autorisation évaluant la sécurité chez des patients atteints de polyarthrite rhumatoïde âgés de 50 ans ou plus et présentant au moins un facteur de risque cardiovasculaire supplémentaire, les taux d’incidence (IC à 95 %) des cancers du poumon pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF étaient respectivement de 0,23 (0,12 – 0,40), de 0,32 (0,18 – 0,51) et de 0,13 (0,05 – 0,26) événements pour 100 patients-années (voir rubriques 4.4 et 5.1). L’étude a nécessité le suivi d’au moins 1 500 patients pendant 3 ans.
Les taux d’incidence (IC à 95 %) des lymphomes pour le tofacitinib 5 mg deux fois par jour, le tofacitinib 10 mg deux fois par jour et les inhibiteurs du TNF étaient respectivement de 0,07 (0,02 – 0,18), de 0,11 (0,04 – 0,24) et de 0,02 (0,00 – 0,10) événements pour 100 patients-années (voir rubriques 4.4 et 5.1).
Population pédiatrique
Arthrite juvénile idiopathique polyarticulaire et RP juvénile
Les effets indésirables observés chez les patients atteints d’AJI dans le cadre du programme de développement clinique étaient cohérents en termes de type et de fréquence avec ceux observés chez les patients adultes atteints de PR, à l’exception de certaines infections (grippe, pharyngite, sinusite, infection virale) et de troubles gastro-intestinaux ou généraux (douleurs abdominales, nausées, vomissements, fièvre, céphalées, toux), qui ont été plus fréquents dans la population pédiatrique atteinte d’AJI. Le MTX était le csDMARD le plus fréquemment utilisé en association (au Jour 1, 156 des 157 patients sous csDMARD ont pris du MTX). Il n’existe pas de données suffisantes concernant le profil de sécurité de tofacitinib utilisé en association avec d’autres csDMARD.
Infections
Dans la phase en double aveugle de l’étude pivot de phase 3 (étude JIA-I), l’infection était l’effet indésirable le plus fréquemment rapporté (44,3 %). Les infections étaient généralement d’intensité légère à modérée.
Dans la population intégrée de sécurité, 7 patients ont présenté des infections graves pendant le traitement par tofacitinib au cours de la période de déclaration (jusqu’à 28 jours après l’administration de la dernière dose du médicament à l’étude), ce qui représente un taux d’incidence de 1,92 patient avec événements pour 100 patient-années : pneumonie, empyème épidural (avec sinusite et abcès sous-périosté), kyste pilonidal, appendicite, pyélonéphrite à Escherichia, abcès de membre et IVU.
Dans la population intégrée de sécurité, 3 patients ont présenté des événements non graves de zona pendant la fenêtre de déclaration, ce qui représente un taux d’incidence de 0,82 patient avec des événements pour 100 patient-années. Un (1) patient supplémentaire a présenté un événement de zona grave en dehors de la fenêtre de déclaration.
Événements hépatiques
Les patients de l’étude pivot dans l’AJI devaient présenter des taux d’ASAT et d’ALAT inférieurs à 1,5 fois la limite supérieure de la normale pour être éligibles au recrutement. Dans la population intégrée de sécurité, 2 patients ont présenté des élévations de l’ALAT ≥ 3 fois la LSN lors de 2 visites consécutives. Aucun de ces événements n’a répondu aux critères de la loi de Hy. Les deux patients ont été sous traitement de fond par MTX et chaque événement s’est résolu après l’arrêt du MTX et l’arrêt définitif de tofacitinib.
Tests de laboratoire
Les modifications des tests de laboratoire chez les patients atteints d’AJI dans le cadre du programme de développement clinique ont été conformes à celles observées chez les patients adultes atteints de PR. Les patients de l’étude pivot sur l’AJI devaient avoir une numération plaquettaire ≥ 100 000 cellules/mm3 pour être éligibles au recrutement ; par conséquent, aucune information n’est disponible pour les patients atteints d’AJI présentant une numération plaquettaire < 100 000 cellules/mm3 avant de commencer le traitement par tofacitinib.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique : Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance
Site internet : www.notifieruneffetindesirable.be
E-mail : adr@fagg-afmps.be
Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santéSite internet : www.guichet.lu/pharmacovigilance.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Pfizer Europe MA EEIG
Boulevard de la Plaine 17
1050 Bruxelles
Belgique
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1178/001
EU/1/17/1178/002
EU/1/17/1178/003
EU/1/17/1178/004
EU/1/17/1178/005
EU/1/17/1178/006
EU/1/17/1178/007
EU/1/17/1178/008
EU/1/17/1178/009
EU/1/17/1178/014
10. DATE DE MISE À JOUR DU TEXTE
09/01/26
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/.
26A09
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3558210 | XELJANZ 5MG COMP PELL 56 X 5MG BLISTER | L04AA29 | € 883,74 | - | Oui | € 12,8 | € 8,5 |
| 3558244 | XELJANZ 5MG COMP PELL 180 X 5MG FLACON | L04AA29 | € 2444,22 | - | Oui | € 15,9 | € 10,5 |
| 3611613 | XELJANZ 5MG COMP PELL 182 X 5MG BLISTER | L04AA29 | € 2471,26 | - | Oui | € 15,9 | € 10,5 |
| 3786415 | XELJANZ 10MG COMP PELL 112 X 10MG BLISTER | L04AA29 | € 2711,88 | - | Oui | € 15,9 | € 10,5 |
| 3786423 | XELJANZ 10MG COMP PELL 56 X 10MG BLISTER | L04AA29 | € 1361,42 | - | Oui | € 12,8 | € 8,5 |
| 3809803 | XELJANZ 5MG COMP PELL 112 X 5MG BLISTER | L04AA29 | € 1361,42 | - | Oui | € 15,9 | € 10,5 |