RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Kevzara 150 mg, solution injectable en seringue préremplie
Kevzara 150 mg, solution injectable en stylo prérempli
Kevzara 200 mg, solution injectable en seringue préremplie
Kevzara 200 mg, solution injectable en stylo prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Kevzara 150 mg solution injectable en seringue préremplie
Chaque seringue préremplie contient 150 mg de sarilumab dans 1,14 ml de solution (131,6 mg/ml).
Kevzara 150 mg, solution injectable en stylo prérempli
Chaque stylo prérempli contient 150 mg de sarilumab dans 1,14 ml de solution (131,6 mg/ml).
Kevzara 200 mg, solution injectable en seringue préremplie
Chaque seringue préremplie contient 200 mg de sarilumab dans 1,14 ml de solution (175 mg/ml).
Kevzara 200 mg, solution injectable en stylo prérempli
Chaque stylo prérempli contient 200 mg de sarilumab dans 1,14 ml de solution (175 mg/ml).
Le sarilumab est un anticorps monoclonal humain, produit par la technologie de l’ADN recombinant dans des cellules ovariennes de hamster chinois.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable
Solution stérile transparente, incolore à jaune pâle, de pH environ 6,0.
Kevzara 150 mg, solution injectable
298-346 mmol/kg
Kevzara 200 mg, solution injectable
306-371 mmol/kg
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Polyarthrite rhumatoïde
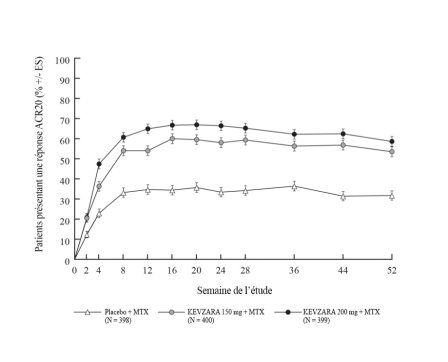
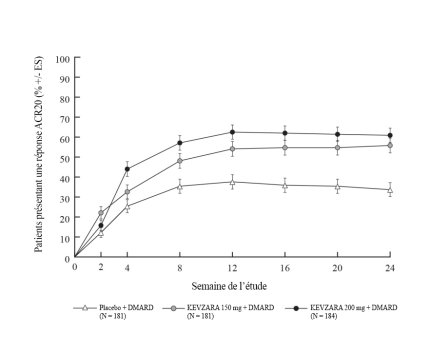
Kevzara est indiqué en association au méthotrexate (MTX) chez les patients adultes atteints de polyarthrite rhumatoïde (PR) active modérée à sévère ayant eu une réponse inadéquate ou intolérants à un ou plusieurs traitements de fond (DMARDs). Kevzara peut être utilisé en monothérapie en cas d’intolérance au MTX ou lorsque le traitement avec le MTX est inadapté (voir rubrique 5.1).
Pseudopolyarthrite rhizomélique
Kevzara est indiqué dans le traitement de la pseudopolyarthrite rhizomélique (PPR) chez les patients adultes ayant eu une réponse inadéquate aux corticoïdes ou ayant présenté une rechute au cours de la diminution progressive des corticoïdes.
4.2 Posologie et mode d’administration
Le traitement doit être instauré et supervisé par des professionnels de santé ayant l’expérience du diagnostic et du traitement de la pathologie pour laquelle ce médicament est destiné (voir rubrique 4.1). Les patients doivent recevoir la carte patient.
Posologie
Polyarthrite rhumatoïde
La posologie recommandée du sarilumab est de 200 mg toutes les 2 semaines administrée en injection sous-cutanée.
Pseudopolyarthrite rhizomélique
La posologie recommandée de sarilumab est de 200 mg une fois toutes les 2 semaines, administrée par injection sous-cutanée, en association avec une diminution progressive des corticoïdes systémiques, après quoi le sarilumab peut être poursuivi en monothérapie.
Les données sont disponibles chez les patients traités jusqu’à 1 an. Le traitement au-delà de 52 semaines doit donc être déterminé selon l’activité de la maladie, la décision du médecin et le choix du patient.
Modification de la posologie :
Polyarthrite rhumatoïde
Une réduction de la posologie de 200 mg une fois toutes les 2 semaines à 150 mg une fois toutes les 2 semaines est recommandée en cas de survenue d’une neutropénie, d’une thrombopénie et d’une élévation des enzymes hépatiques.
Le traitement par le sarilumab doit être interrompu chez les patients qui développent une infection grave et ce jusqu’à ce que cette infection soit contrôlée.
L’instauration d’un traitement par le sarilumab n’est pas recommandée chez les patients présentant un nombre de neutrophiles bas, c’est-à-dire un nombre absolu de neutrophiles (NAN) inférieur à 2 000/mm3.
L’instauration d’un traitement par le sarilumab n’est pas recommandée chez les patients présentant un nombre de plaquettes inférieur à 150 000/mm3.
Tableau 1 : Modifications de la posologie recommandées en cas de neutropénie, de thrombopénie ou d’élévation des enzymes hépatiques dans la polyarthrite rhumatoïde (voir rubriques 4.4 et 4.8) :
Nombre absolu de neutrophiles bas (voir rubrique 5.1) | |
Paramètre biologique (cellules/mm3) | Recommandation |
NAN supérieur à 1000 | Conserver la posologie du sarilumab existante. |
NAN entre 500 et 1 000 | Interrompre le traitement par le sarilumab jusqu’à ce que la valeur soit > 1 000/mm3. |
NAN inférieure à 500 | Arrêter le traitement par le sarilumab. |
Nombre de plaquettes bas | |
Paramètre biologique (cellules x 103/mm3) | Recommandation |
Entre 50 et 100 | Interrompre le traitement par le sarilumab jusqu’à ce que la valeur soit > 100 x 103/mm3. |
Inférieure à 50 | Après confirmation par des examens répétés, arrêter le traitement par le sarilumab. |
Anomalies des enzymes hépatiques | |
Paramètre biologique | Recommandation |
ALAT > 1 x et ≤ 3 x la limite supérieure de la normale (LSN) | Envisager une modification de la posologie des DMARDs associés en fonction de l’état clinique ou des agents immunomodulateurs. |
ALAT > 3 x et ≤ 5 x LSN | Interrompre le traitement par le sarilumab jusqu’à ce que la valeur soit < 3 x LSN. |
ALAT > 5 x LSN | Arrêter le traitement par le sarilumab. |
Pseudopolyarthrite rhizomélique (PPR)
Anomalies biologiques : Arrêt du traitement par sarilumab chez les patients atteints de PPR qui développent les anomalies biologiques suivantes (voir rubriques 4.4 et 5.1) :
- neutropénie (NAN inférieur à 1 x 109/L à la fin de l’intervalle entre les prises)
- thrombopénie (numération plaquettaire inférieure à 100 x 103 μL)
- augmentation des ASAT ou des ALAT (3 fois au-dessus de la LSN)
Les modifications de posologie n’ont pas été étudiées chez les patients atteints de PPR présentant ces anomalies. Pour les critères d’initiation du traitement, se référer à la posologie de la PPR.
Dose oubliée
En cas d’oubli d’une injection du sarilumab, si l’oubli est constaté dans les 3 jours, l’injection doit être réalisée immédiatement. L’injection suivante devra être réalisée à la date initialement prévue.
Si l’oubli est de 4 jours ou plus, l’injection devra être réalisée à la date prévue de l’injection suivante sans doubler la dose.
Populations spéciales
Insuffisance rénale
Aucun ajustement de la posologie n’est nécessaire chez les patients ayant une insuffisance rénale légère à modérée. Le sarilumab n’a pas été étudié chez les patients présentant une insuffisance rénale sévère (voir rubrique 5.2).
Insuffisance hépatique
La sécurité et l’efficacité du sarilumab n’ont pas été étudiées chez les patients ayant une insuffisance hépatique, y compris chez les patients présentant une sérologie positive au virus de l’hépatite B (VHB) ou de l’hépatite C (VHC) (voir rubrique 4.4).
Personnes âgées
Aucun ajustement de la posologie n’est nécessaire chez les patients âgés de plus de 65 ans (voir rubrique 4.4).
Population pédiatrique
La sécurité et l’efficacité de la seringue préremplie et du stylo prérempli de sarilumab chez les enfants de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Utilisation par voie sous-cutanée.
Les sites d’injection (abdomen, cuisse et bras) doivent varier à chaque injection. Le sarilumab ne doit pas être injecté dans une peau sensible, lésée ou présentant des ecchymoses ou des cicatrices.
Seringue préremplie et stylo prérempli
L’intégralité du contenu (1,14 ml) de la seringue préremplie ou du stylo prérempli doit être administrée par injection sous-cutanée.
Pour la seringue préremplie/le stylo prérempli, l’injection du sarilumab peut être effectuée par le patient lui-même ou par un aidant si le professionnel de santé considère cela approprié. Une formation appropriée à la préparation et à l’administration du sarilumab doit être dispensée aux patients et/ou aux aidants avant utilisation.
La seringue préremplie ou le stylo n’ont pas été étudiés chez les patients pédiatriques.
Des instructions complètes pour l'administration de ce médicament sont données dans la notice.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Infections sévères actives (voir rubrique 4.4).
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquents dans la PR (n=661) et la PPR (n=59) sont : neutropénies (14,3%), infections des voies aériennes supérieures (6,8%), augmentation des ALAT (6,3%), infections des voies urinaires (5,3%) et érythème au site d’injection (5,0%). Les effets indésirables graves les plus fréquents sont les infections (3,1%) (voir rubrique 4.4).
Tableau des effets indésirables
Les effets indésirables listés dans ce tableau ont été rapportés au cours des études cliniques contrôlées. La fréquence des effets indésirables énumérés ci-dessous est définie selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). À l’intérieur de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 : Effets Indésirables chez les patients atteints de PR et PPR
Classe de systèmes d’organes MedDRA | Fréquence | Effet indésirable |
Infections et infestations | Fréquent | Infection des voies aériennes supérieures |
Infection des voies urinaires | ||
Herpès buccal | ||
Cellulite | ||
Pneumonie | ||
Peu fréquent | Rhinopharyngite | |
| ||
Affections hématologiques et du système lymphatique | Très fréquent | Neutropénie* |
Fréquent | Leucopénie* | |
Thrombopénie | ||
Troubles du métabolisme et de la nutrition | Fréquent | Hypertriglycéridémie |
Hypercholestérolémie | ||
Affections gastro-intestinales | Rare | Perforation gastro-intestinale |
Affections hépatobiliaires | Fréquent | Transaminases augmentées |
Troubles généraux et anomalies au site d’administration | Fréquent | Érythème au site d’injection |
Prurit au site d’injection* |
* Dans l’étude SAPHYR, les effets indésirables rapportés chez les patients atteints de PPR sont la neutropénie, la leucopénie et le prurit au site d’injection.
Description de certains effets indésirables
Polyarthrite rhumatoïde
Infections
Dans la population contrôlée versus placebo, les taux d’infections ont été de 84,5 ; 81,0 et 75,1 événements pour 100 patients-années, dans les groupes sarilumab 200 mg + DMARDs, sarilumab 150 mg + DMARDs et placebo + DMARDs, respectivement. Les infections les plus fréquemment rapportées (5 à 7 % des patients) étaient des infections des voies aériennes supérieures, des infections des voies urinaires et des rhinopharyngites. Les taux d’infections graves ont été de 4,3 ; 3,0 et 3,1 événements pour 100 patients-années, dans les groupes sarilumab 200 mg + DMARDs, sarilumab 150 mg + DMARDs et placebo + DMARDs, respectivement.
Dans la population de sécurité long terme sarilumab + DMARDs, les taux d’infections et d’infections graves ont été de 57,3 et 3,4 événements pour 100 patients-années, respectivement.
Les infections graves les plus fréquemment observées comprenaient la pneumonie et la cellulite. Des cas d’infections opportunistes ont été rapportés (voir rubrique 4.4).
Les taux globaux d’infections et d’infections graves dans la population recevant le sarilumab en monothérapie ont été cohérents avec les taux obtenus dans la population sarilumab + DMARDs.
Perforation gastro-intestinale
Des cas de perforation gastro-intestinale ont été rapportés chez des patients présentant ou non une diverticulite. La plupart des patients ayant développé des perforations gastro-intestinales prenaient en association des anti-inflammatoires non stéroïdiens (AINS), des corticostéroïdes ou du MTX. La contribution relative de ces médicaments pris en association avec le sarilumab dans le développement de perforations gastro-intestinales n’est pas connue (voir rubrique 4.4).
Réactions d’hypersensibilité
Dans les études contrôlées versus placebo, la proportion de patients pour lesquels le traitement a été interrompu en raison de réactions d’hypersensibilité a été supérieure chez ceux traités par sarilumab (0,9 % dans le groupe 200 mg, 0,5 % dans le groupe 150 mg) par rapport à ceux recevant le placebo (0,2 %). Les taux d’interruptions en raison d’une hypersensibilité dans la population étudiée pour la sécurité à long terme sarilumab + DMARDs et celle recevant sarilumab en monothérapie ont été comparables à ceux de la population contrôlée versus placebo. Dans la population contrôlée versus placebo, des effets indésirables graves consistant en des réactions d’hypersensibilité ont été rapportés chez 0,2 % des patients traités par sarilumab 200 mg toutes les 2 semaines + DMARDs. Aucun cas n’a été rapporté dans le groupe sarilumab 150 mg toutes les 2 semaines + DMARDs.
Réactions au site d’injection
Dans les études contrôlées versus placebo, des réactions au site d’injection ont été rapportées chez 9,5 %, 8 % et 1,4 % des patients recevant sarilumab 200 mg, sarilumab 150 mg et le placebo, respectivement. Ces réactions au site d’injection (comprenant érythème et prurit) ont été de sévérité légère à modérée chez la majorité des patients (99,5%, 100% et 100% chez sarilumab 200 mg, sarilumab 150 mg et le placebo, respectivement). Chez deux patients sous sarilumab (0,2 %), le traitement a été interrompu en raison de réactions au site d’injection.
Anomalies des paramètres biologiques
Pour permettre une comparaison directe de la fréquence des anomalies des paramètres biologiques entre le groupe recevant le placebo et celui recevant le traitement actif, les données utilisées ont été celles des semaines 0 à 12, période qui a précédé la possibilité pour les patients de passer du placebo au sarilumab.
Nombre de neutrophiles
Des diminutions du nombre de neutrophiles en dessous d’une valeur de 1 000/mm3 ont été rapportées chez 6,4 % et 3,6 % des patients des groupes sarilumab 200 mg + DMARDs et sarilumab 150 mg + DMARDs, respectivement. Aucune n’a été rapportée dans le groupe placebo + DMARDs. Des diminutions du nombre de neutrophiles en dessous d’une valeur de 500/mm3 ont été rapportées chez 0,8 % et 0,6 % des patients dans les groupes sarilumab 200 mg + DMARDs et sarilumab 150 mg + DMARDs, respectivement. Chez les patients ayant une diminution du nombre absolu de neutrophiles (NAN), une modification du schéma thérapeutique comme une interruption du sarilumab ou une diminution de la posologie a entraîné une augmentation ou une normalisation du NAN (voir rubrique 4.2). La réduction du NAN n’a pas été associée à une augmentation de l’incidence des infections, y compris des infections graves.
Dans la population de sécurité long terme sarilumab + DMARDs et celle recevant sarilumab en monothérapie, les observations sur la numération des neutrophiles ont été cohérentes avec celles de la population contrôlée versus placebo (voir rubrique 4.4).
Nombre de plaquettes
Une diminution du nombre de plaquettes en dessous d’une valeur de 100 x 103/mm3 a été rapportée chez 1,2 % et 0,6 % des patients dans les groupes sarilumab200 mg + DMARDs et sarilumab 150 mg + DMARDs, respectivement. Aucune n’a été rapportée dans le groupe placebo + DMARDs.
Dans la population étudiée pour la sécurité à long terme sarilumab + DMARDs et la population recevant sarilumab en monothérapie, les observations sur la numération plaquettaire ont été cohérentes avec celles de la population contrôlée versus placebo.
Aucun événement hémorragique n’a été associé aux diminutions du nombre de plaquettes.
Enzymes hépatiques
Les anomalies des enzymes hépatiques sont résumées dans le Tableau 3. Chez les patients présentant une élévation des enzymes hépatiques, une modification du schéma thérapeutique telle qu’une interruption du traitement ou une réduction de la posologie a entraîné une diminution ou une normalisation des enzymes hépatiques (voir rubrique 4.2). Ces élévations n’ont pas été associées à une élévation cliniquement significative de la bilirubine conjuguée, ni à des signes cliniques d’hépatite ou d’insuffisance hépatique (voir rubrique 4.4).
Tableau 3 : Incidence des anomalies des enzymes hépatiques dans les études cliniques contrôlées
| Placebo + DMARD | Sarilumab 150 mg + DMARD | Sarilumab 200 mg + DMARD | Sarilumab en monothérapie, toute dose |
ASAT |
|
|
|
|
> 3 x LSN – | 0 % | 1,2 % | 1,1 % | 1,1 % |
> 5 x LSN | 0 % | 0,6 % | 0,2 % | 0 % |
ALAT |
|
|
|
|
> 3 x LSN – | 0,6 % | 3,2 % | 2,4 % | 1,9 % |
> 5 x LSN | 0 % | 1,1 % | 0,8 % | 0,2 % |
Lipides
Les paramètres lipidiques (LDL, HDL et triglycérides) ont été évalués initialement 4 semaines après l’instauration du traitement par sarilumab + DMARDs dans la population contrôlée versus placebo. À la semaine 4, le taux moyen de LDL montrait une augmentation de 14 mg/dl, le taux moyen de triglycérides, une augmentation de 23 mg/dl, et le taux moyen de HDL, une augmentation de 3 mg/dl. Après la semaine 4, aucune nouvelle augmentation n’a été observée. Aucune différence significative n’a été relevée entre les doses.
Dans la population étudiée pour la sécurité à long terme sarilumab + DMARDs et la population recevant sarilumab en monothérapie, les observations sur les paramètres lipidiques ont été cohérentes avec celles de la population contrôlée versus placebo.
Tumeurs malignes
Dans la population de l’étude contrôlée contre placebo, les tumeurs malignes sont survenues à la même fréquence chez les patients recevant soit le sarilumab + DMARDs soit le placebo + DMARDs (1,0 événement pour 100 patients-années).
Dans la population de tolérance à long terme du sarilumab + DMARDs et dans la population recevant le sarilumab en monothérapie, les taux de tumeurs malignes étaient cohérents avec les taux observés dans la population de l’étude contrôlée contre placebo (voir rubrique 4.4).
Immunogénicité
Comme toutes les protéines thérapeutiques, le sarilumab possède un potentiel d’immunogénicité.
Dans la population contrôlée versus placebo, 4,0 %, 5,6 % et 2,0 % des patients traités par sarilumab 200 mg + DMARDs, sarilumab 150 mg + DMARDs et placebo + DMARDs respectivement, avaient eu des anticorps anti-médicaments (anti-drug antibody, ADA). Des anticorps neutralisants (neutralizing antibody, NAb) ont été détectés chez 1,0 %, 1,6 % et 0,2 % des patients sous sarilumab 200 mg, sarilumab 150 mg et placebo, respectivement.
Dans la population recevant sarilumab en monothérapie, les observations ont été comparables à celles faites dans la population sarilumab + DMARDs.
La formation d’anticorps anti-médicament (ADA) est susceptible de modifier la pharmacocinétique du sarilumab. Aucune corrélation n’a été observée entre la formation d’anticorps anti-sarilumab et une perte d’efficacité ou la survenue d’effets indésirables.
Pseudopolyarthrite rhizomélique
La tolérance du sarilumab a été étudiée au cours d’une étude de phase 3 (SAPHYR) chez 117 patients atteints de PPR, dont 59 ont reçu du sarilumab 200 mg par voie sous-cutanée (voir rubrique 5.1). La durée totale des patients-années dans la population atteinte de PPR était de 47,37 patients-années au cours de l’étude en double aveugle de 12 mois, contrôlée versus placebo. Les données de tolérance sont disponibles jusqu’à 1 an.
Infections
Dans l’étude SAPHYR, la proportion de patients présentant des infections était plus faible dans le groupe sarilumab 200 mg avec diminution progressive de la prednisone sur 14 semaines (37,3 %) que dans le groupe placebo avec diminution progressive de la prednisone sur 52 semaines (50,0 %). Des infections graves ont été rapportées chez 3 (5,1 %) patients dans le groupe sarilumab 200 mg avec diminution progressive de la prednisone sur 14 semaines (tous étaient des cas d’infections bactériennes) et chez 3 (5,2 %) patients dans le groupe placebo avec diminution progressive de la prednisone sur 52 semaines (tous les cas étaient des infections par la COVID-19).
Anomalies biologiques
Numération des neutrophiles
Dans l’étude SAPHYR, des diminutions du nombre de neutrophiles en dessous de 1 x 109/L sont survenues chez 7 (12 %) patients du groupe sarilumab, dont 2 (3,4 %) étaient graves (diminution du nombre de neutrophiles en dessous de 0,5 x 109/ L).
Enzymes hépatiques
Dans l’étude SAPHYR, aucun patient traité par le sarilumab n’a présenté d’ALAT ou d’ASAT supérieure à 3 fois la limite supérieure de la normale (LSN). Dans le groupe placebo, 2 patients ont présenté une élévation des ALAT supérieure à 3 fois la limite supérieure de la normale.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe un risque potentiel d’immunogénicité avec le sarilumab.
Dans la population PPR, 1 (1,8 %) patient traité par 200 mg de sarilumab a présenté une réponse persistante en anticorps anti-médicament (ADA) et aucun des patients du groupe placebo n’a présenté de réponse en ADA. Une réponse positive au test des anticorps neutralisants a été détectée chez les patients atteints de PPR présentant une réponse en ADA sous sarilumab 200 mg. En raison de la faible fréquence des ADA, l’effet de ces anticorps sur la sécurité d’emploi et/ou l’efficacité du sarilumab n’est pas connu.
Population pédiatrique
La sécurité et l’efficacité de la seringue préremplie et du stylo prérempli de sarilumab chez les enfants de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique : Agence Fédérale des Médicaments et des Produits de Santé : www.afmps.be – Division Vigilance : Site internet : www.notifieruneffetindesirable.be – E-mail : adr@fagg-afmps.be
Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé – Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Sanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
France
8. NUMÉROS D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1196/001
EU/1/17/1196/002
EU/1/17/1196/003
EU/1/17/1196/004
EU/1/17/1196/005
EU/1/17/1196/006
EU/1/17/1196/007
EU/1/17/1196/008
EU/1/17/1196/009
EU/1/17/1196/010
EU/1/17/1196/011
EU/1/17/1196/012
10. DATE DE MISE À JOUR DU TEXTE
02/2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3593662 | KEVZARA 150MG SOL INJ STYLO PREREMP.VERRE 2X1,14ML | L04AC14 | € 923,67 | - | Oui | € 12,8 | € 8,5 |
| 3593670 | KEVZARA 200MG SOL INJ STYLO PREREMP.VERRE 2X1,14ML | L04AC14 | € 923,67 | - | Oui | € 12,8 | € 8,5 |
| 3593688 | KEVZARA 200MG SOL INJ SER PRERREMPL.VERRE 2X1,14ML | L04AC14 | € 923,67 | - | Oui | € 12,8 | € 8,5 |
| 3593696 | KEVZARA 150MG SOL INJ SER PREREMPL.VERRE 2X1,14ML | L04AC14 | € 923,67 | - | Oui | € 12,8 | € 8,5 |