RESUME DES CARACTERISTIQUES DU PRODUIT
1. DENOMINATION DU MEDICAMENT
Forxiga 5 mg, comprimés pelliculés
Forxiga 10 mg, comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Forxiga 5 mg, comprimés pelliculés
Chaque comprimé contient du propylène glycol monohydraté de dapagliflozine équivalent à 5 mg de dapagliflozine.
Excipient à effet notoire
Chaque comprimé de 5 mg contient 25 mg de lactose.
Forxiga 10 mg, comprimés pelliculés
Chaque comprimé contient du propylène glycol monohydraté de dapagliflozine équivalent à 10 mg de dapagliflozine.
Excipient à effet notoire
Chaque comprimé de 10 mg contient 50 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé).
Forxiga 5 mg, comprimés pelliculés
Comprimés pelliculés, jaunes, biconvexes, ronds, d’un diamètre de 0,7 cm avec « 5 » gravé sur une face et « 1427 » gravé sur l’autre face.
Forxiga 10 mg, comprimés pelliculés
Comprimés pelliculés, jaunes, biconvexes, en forme de losange, d’approximativement 1,1 x 0,8 cm de diagonale, avec « 10 » gravé sur une face et « 1428 » gravé sur l’autre face.
4. DONNEES CLINIQUES
4.1 Indications thérapeutiques
Diabète de type 2
Forxiga est indiqué chez les adultes et chez les enfants de 10 ans et plus pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d’un régime alimentaire et de l’exercice physique :
- en monothérapie quand la metformine est considérée comme inappropriée en raison d’une intolérance.
- en plus d’autres médicaments destinés au traitement du diabète de type 2.
Voir rubriques 4.4, 4.5 et 5.1 pour les résultats des études concernant les associations de traitements, les effets sur le contrôle glycémique, les événements cardiovasculaires et rénaux, et les populations étudiées.
Insuffisance cardiaque
Forxiga est indiqué chez les adultes pour le traitement de l’insuffisance cardiaque chronique symptomatique.
Maladie rénale chronique
Forxiga est indiqué chez les adultes pour le traitement de la maladie rénale chronique.
4.2 Posologie et mode d’administration
Posologie
Diabète de type 2
La dose recommandée est 10 mg de dapagliflozine une fois par jour.
Lorsque la dapagliflozine est utilisée en association avec l’insuline ou un sécrétagogue d’insuline, comme les sulfamides hypoglycémiants, une dose plus faible d’insuline ou d’un sécrétagogue d’insuline peut être envisagée pour réduire le risque d’hypoglycémie (voir rubriques 4.5 et 4.8).
Insuffisance cardiaque
La dose recommandée est 10 mg de dapagliflozine une fois par jour.
Maladie rénale chronique
La dose recommandée est 10 mg de dapagliflozine une fois par jour.
Populations particulières
Insuffisance rénale
Aucun ajustement de la dose n’est nécessaire selon l’état de la fonction rénale.
En raison de l'expérience limitée, il n’est pas recommandé d'initier un traitement par la dapagliflozine chez les patients avec un DFG < 25 mL/min.
Chez les patients diabétiques de type 2, l’efficacité glycémique de la dapagliflozine est réduite lorsque le débit de filtration glomérulaire (DFG) est < 45 mL/min et est vraisemblablement absente chez les patients atteints d’insuffisance rénale sévère. Par conséquent, si le DFG diminue au-dessous de 45 mL/min, un traitement hypoglycémiant supplémentaire doit être envisagé chez les patients diabétiques de type 2 si un contrôle glycémique complémentaire est nécessaire (voir rubriques 4.4, 4.8, 5.1 et 5.2).
Insuffisance hépatique
Aucun ajustement de la dose n’est nécessaire chez les patients atteints d’insuffisance hépatique légère ou modérée. Chez les patients atteints d’insuffisance hépatique sévère, la dose initiale recommandée est 5 mg. Si le traitement est bien toléré, la dose peut être augmentée à 10 mg (voir rubriques 4.4 et 5.2).
Sujets âgés (≥ 65 ans)
Aucun ajustement de la dose n’est recommandé selon l’âge.
Population pédiatrique
Aucun ajustement de la dose n’est nécessaire pour le traitement du diabète de type 2 chez les enfants âgés de 10 ans et plus (voir rubriques 5.1 et 5.2). Aucune donné n’est disponible chez les enfants en-dessous de 10 ans.
La tolérance et l’efficacité de dapagliflozine pour le traitement de l’insuffisance cardiaque ou le traitement de la maladie rénale chronique chez les enfants âgés < 18 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
Forxiga peut être pris par voie orale, une fois par jour, à tout moment de la journée, au cours ou en dehors des repas. Les comprimés doivent être avalés entiers.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
Diabète de type 2
Dans les études cliniques conduites dans le diabète de type 2, plus de 15 000 patients ont été traités par dapagliflozine.
L’évaluation principale de sécurité d’emploi et de tolérance a été réalisée dans le cadre d’une analyse poolée préspécifiée de 13 études à court terme (jusqu’à 24 semaines) contrôlées versus placebo avec 2 360 patients traités par dapagliflozine 10 mg et 2 295 par placebo.
Dans l’étude des effets cardiovasculaires conduite avec la dapagliflozine, dans le diabète de type 2 (étude DECLARE voir rubrique 5.1), 8 574 patients ont reçu de la dapagliflozine 10 mg et 8 569 ont reçu un placebo pendant une durée d’exposition médiane de 48 mois. En tout, il y a eu 30 623 patients-années d’exposition à la dapagliflozine.
Les effets indésirables les plus fréquemment rapportés dans les études cliniques étaient les infections génitales.
Insuffisance cardiaque
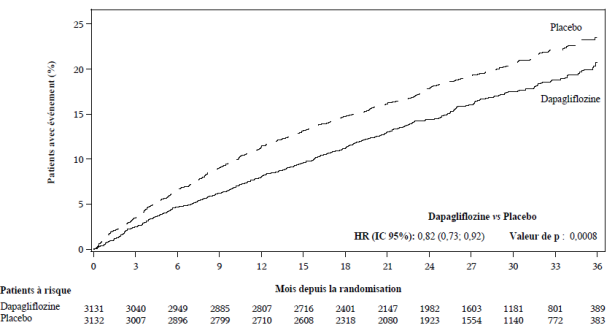
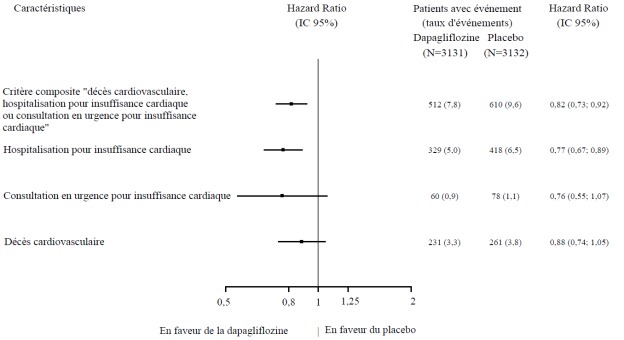
Dans l’étude des effets cardiovasculaires conduite avec la dapagliflozine chez des patients atteints d’insuffisance cardiaque à fraction d’éjection réduite (étude DAPA-HF), 2 368 patients ont été traités par la dapagliflozine à la dose de 10 mg et 2 368 patients ont reçu un placebo pendant une durée d’exposition médiane de 18 mois. La population de patients incluait des patients diabétiques de type 2 ou non diabétiques et des patients avec un DFGe ≥ 30 mL/min/1,73 m2. Dans l’étude des effets cardiovasculaires conduite avec la dapagliflozine chez des patients atteints d’insuffisance cardiaque avec une fraction d’éjection ventriculaire gauche > 40 % (DELIVER), 3 126 patients ont été traités par la dapagliflozine à la dose de 10 mg et 3 127 patients ont reçu un placebo pendant une durée d’exposition médiane de 27 mois. La population de patients incluait des patients diabétiques de type 2 ou non diabétiques et des patients avec un DFGe ≥ 25 mL/min/1,73 m2.
Le profil de sécurité global de la dapagliflozine chez les patients atteints d’insuffisance cardiaque était cohérent avec le profil de sécurité connu de la dapagliflozine.
Maladie rénale chronique
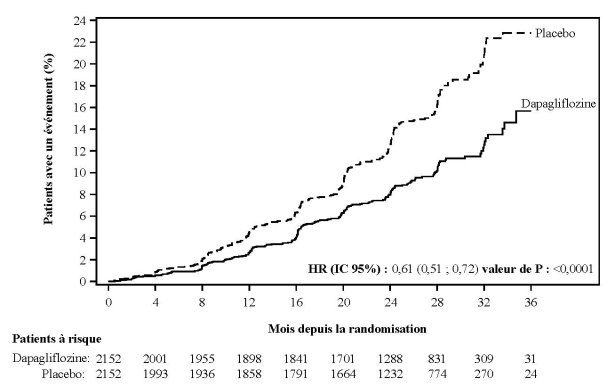
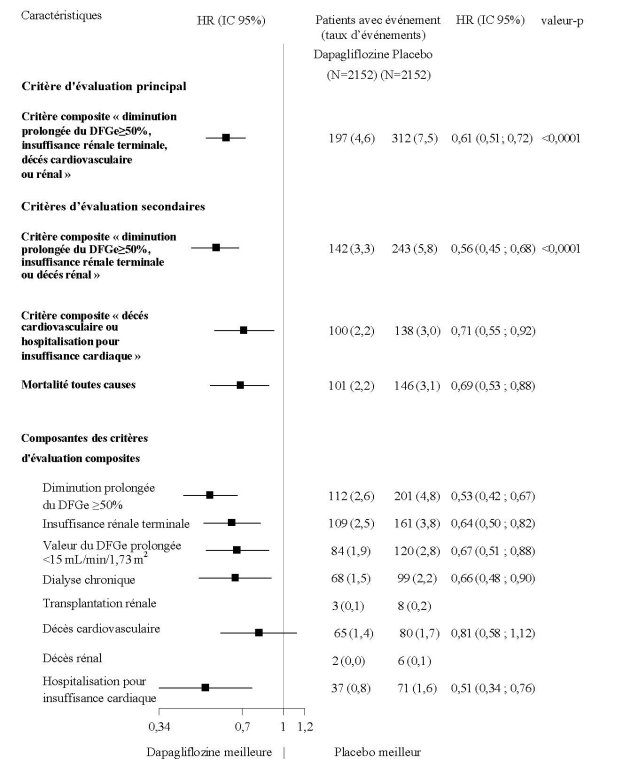
Dans l’étude des effets rénaux conduite avec la dapagliflozine chez des patients atteints de maladie rénale chronique (DAPA-CKD), 2 149 patients ont été traités par la dapagliflozine à la dose de 10 mg et 2 149 patients ont reçu un placebo pendant une durée d’exposition médiane de 27 mois. La population de patients incluait des patients diabétiques de type 2 et non diabétiques, avec un DFGe ≥ 25 à ≤ 75 mL/min/1,73 m², et une albuminurie (rapport albuminurie/créatininurie [RAC] compris entre 200 et 5000 mg/g). Le traitement était poursuivi si le DFGe diminuait à moins de 25 mL/min/1,73 m².
Le profil de sécurité global de la dapagliflozine chez les patients atteints de maladie rénale chronique était cohérent avec le profil de sécurité connu de la dapagliflozine.
Liste tabulée des effets indésirables
Les effets indésirables suivants ont été identifiés dans les études cliniques contrôlées versus placebo et lors de la surveillance en post-commercialisation. Aucun ne s’est révélé dose‑dépendant. Les effets indésirables mentionnés ci‑dessous sont classés par fréquence et par classe de systèmes d’organes (SOC). Les différentes catégories de fréquence adoptent la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1. Effets indésirables issus d’études cliniques contrôlées versus placeboa et de l’expérience post-commercialisation
Classe de systèmes d’organes | Très fréquent | Fréquent* | Peu fréquent** | Rare | Très rare |
Infections et infestations |
| Vulvovaginite, balanite et infections génitales associées*,b,c | |
| Fasciite nécrosante du périnée (gangrène de Fournier)b,i |
Troubles du métabolisme et de la nutrition | Hypoglycémie (quand utilisé avec SU ou insuline)b |
| Déplétion volémiqueb,e | Acidocétose diabétique (dans le cadre d’une utilisation dans le diabète de type 2)b,i,k |
|
Affections du système nerveux |
| Sensations vertigineuses |
|
|
|
Affections gastro‑intestinales |
|
| Constipation** |
|
|
Affections de la peau et du tissu sous-cutané |
| Rashj |
|
| Angio-œdème |
Affections musculo‑squelettiques et systémiques |
| Douleur dorsale* |
|
|
|
Affections du rein et des voies urinaires |
| Dysurie | Nycturie** |
| Néphrite tubulo-interstitielle |
Affections des organes de reproduction et du sein |
|
| Prurit vulvo vaginal** |
|
|
Investigations |
| Augmentation de l’hématocriteg | Elévation de la créatininémie pendant le traitement initial**,b |
|
|
a Le tableau présente des données recueillies sur 24 semaines (court terme), n’excluant pas l’administration d’un traitement antidiabétique de secours.
b Voir paragraphe correspondant ci‑dessous pour plus d’informations.
c La vulvovaginite, la balanite et les infections génitales associées incluent, par exemple les termes recommandés prédéfinis : infection mycosique vulvo‑vaginale, infection vaginale, balanite, infection génitale fongique, candidose vulvo‑vaginale, vulvovaginite, balanite candidosique, candidose génitale, infection génitale, infection génitale masculine, infection pénienne, vulvite, vaginite bactérienne, abcès vulvaire.
d L’infection des voies urinaires inclut les termes préférés suivants, mentionnés par ordre de fréquence rapportée : infection des voies urinaires, cystite, infection des voies urinaires par Escherichia, infection des voies génito-urinaires, pyélonéphrite, trigonite, uréthrite, infection rénale et prostatite.
e La déplétion volémique regroupe, par exemple, les termes recommandés prédéfinis suivants : déshydratation, hypovolémie, hypotension.
f La polyurie regroupe les termes préférés suivants : pollakiurie, polyurie, augmentation du volume urinaire
g Les variations moyennes par rapport à la valeur initiale de l’hématocrite étaient 2,30 % pour dapagliflozine 10 mg versus ‑0,33 % pour le placebo. Des valeurs de l’hématocrite >55 % ont été rapportées chez 1,3 % des sujets traités par dapagliflozine 10 mg versus 0,4 % des sujets recevant le placebo.
h La variation moyenne en pourcentage par rapport à la valeur initiale pour la dapagliflozine 10 mg versus placebo, respectivement, était : cholestérol total 2,5 % versus 0,0 % ; HDL cholestérol 6,0 % versus 2,7 % ; LDL cholestérol 2,9 % versus -1,0 % ; triglycérides -2,7 % versus -0,7 %.
i Voir la rubrique 4.4
j L’effet indésirable a été identifié lors de la surveillance en post-commercialisation. Rash inclut les termes préférés suivants, listés par ordre de fréquence dans les études cliniques : rash, rash généralisé, éruption prurigineuse, rash maculeux, rash maculopapuleux, rash pustuleux, rash vésiculeux, et rash érythémateux. Dans les études cliniques contrôlées versus placebo et versus substance active (dapagliflozine, N = 5936, l’ensemble des bras contrôles, N = 3403), la fréquence du rash était similaire pour la dapagliflozine (1,4%) et pour les bras contrôles (1,4 %) respectivement.
k Rapportée dans le cadre de l’étude des effets cardiovasculaires conduite chez des patients atteints de diabète de type 2 (DECLARE). La fréquence est basée sur le taux annuel.
* Rapportés chez ≥ 2 % des sujets et chez ≥ 1 % des sujets avec au moins 3 sujets de plus dans le groupe traité par la dapagliflozine 10 mg par rapport au groupe placebo.
** Rapportés par l’investigateur comme possiblement relié, probablement relié ou relié au traitement de l’étude et rapportés chez ≥ 0,2 % chez des sujets et ≥ 0,1 % chez au moins 3 sujets de plus dans le groupe traité par dapagliflozine 10 mg par rapport au groupe placebo.
Description de certains effets indésirables
Vulvovaginite, balanite et infections génitales associées
Dans l’analyse poolée de 13 études visant à analyser la tolérance, des cas de vulvovaginite, de balanite et d’infections génitales associées ont été rapportés respectivement chez 5,5 % et 0,6 % des patients ayant reçu la dapagliflozine 10 mg et le placebo. La plupart des infections étaient légères à modérées et les patients ont répondu à un traitement standard initial et ont rarement arrêté le traitement par dapagliflozine. Ces infections ont été plus fréquentes chez les femmes (8,4 % et 1,2 % pour la dapagliflozine et le placebo, respectivement) et les patients avec un antécédent étaient plus susceptibles d’avoir une infection récurrente.
Dans l’étude DECLARE, les nombres de patients présentant des événements indésirables graves de type infections génitales étaient faibles et équilibrés : 2 patients dans chacun des groupes dapagliflozine et placebo.
Dans l’étude DAPA-HF, aucun patient n’a rapporté d’événement indésirable grave de type infections génitales dans le groupe dapagliflozine et un patient en a rapporté dans le groupe placebo. Sept (0,3 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo. Dans l’étude DELIVER, un (< 0,1 %) patient dans chaque groupe de traitement a rapporté un événement indésirable grave de type infections génitales. Trois (0,1 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo.
Dans l’étude DAPA-CKD, 3 (0,1 %) patients ont présenté des événements indésirables graves de type infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo. Trois (0,1 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo. Aucun événement indésirable grave de type infections génitales et aucun arrêt du traitement en raison d’infections génitales n’ont été rapportés chez des patients non diabétiques.
Des cas de phimosis/phimosis acquis ont été rapportés concomitamment à des infections génitales et, dans certains cas, une circoncision a été nécessaire.
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de gangrène de Fournier ont été rapportés en post-commercialisation chez des patients prenant des inhibiteurs de SGLT2, incluant la dapagliflozine (voir rubrique 4.4).
Dans l’étude DECLARE conduite chez 17 160 patients avec un diabète de type 2 et avec un temps médian d’exposition de 48 mois, un total de 6 cas de gangrène de Fournier ont été rapportés, un dans le groupe traité par la dapagliflozine et 5 dans le groupe placebo.
Hypoglycémie
La fréquence de l’hypoglycémie dépendait du type de traitement initial utilisé dans les études cliniques dans le diabète.
Pour les études de la dapagliflozine en monothérapie, en association à la metformine ou en association à la sitagliptine (avec ou sans metformine), la fréquence des épisodes mineurs d’hypoglycémie s’est avérée similaire (< 5 %) entre les groupes de traitement, y compris le placebo jusqu’à 102 semaines de traitement. Dans toutes les études, les événements majeurs d’hypoglycémie ont été peu fréquents et comparables entre les groupes traités par la dapagliflozine ou le placebo. Les études en association aux sulfamides hypoglycémiants et aux traitements par insuline avaient des taux plus élevés d’hypoglycémie (voir rubrique 4.5).
Dans une étude en association au glimépiride, aux semaines 24 et 48, des épisodes mineurs d’hypoglycémie ont été rapportés plus fréquemment dans le groupe traité par dapagliflozine 10 mg et glimépiride (6,0 % et 7,9 %, respectivement) que chez les patients ayant reçu le placebo et le glimépiride (2,1 % et 2,1 %, respectivement).
Dans une étude en association à l’insuline, des épisodes d’hypoglycémie majeure ont été rapportés, respectivement aux semaines 24 et 104, chez 0,5 % et 1,0 % du groupe de patients traités par dapagliflozine 10 mg et insuline, et chez 0,5 % du groupe de patients traités par placebo et insuline aux semaines 24 et 104. Aux semaines 24 et 104, des épisodes mineurs d’hypoglycémie ont été rapportés respectivement chez 40,3 % et 53,1 % des patients ayant reçu dapagliflozine 10 mg et insuline et chez 34,0 % et 41,6 % des patients ayant reçu le placebo et insuline.
Dans une étude en association à la metformine et à un sulfamide hypoglycémiant conduite jusqu’à 24 semaines, aucun épisode d’hypoglycémie majeure n’a été rapporté. Des épisodes mineurs d’hypoglycémie ont été rapportés chez 12,8 % des sujets qui ont reçu la dapagliflozine 10 mg plus metformine et un sulfamide hypoglycémiant et chez 3,7 % des sujets qui ont reçu un placebo plus metformine et un sulfamide hypoglycémiant.
Dans l’étude DECLARE, aucune augmentation du risque d’hypoglycémie majeure n’a été observée avec le traitement par dapagliflozine par rapport au placebo. Des événements majeurs d’hypoglycémie ont été rapportés chez 58 (0,7 %) patients traités par dapagliflozine et chez 83 (1,0 %) patients traités par placebo.
Dans l’étude DAPA-HF, des événements majeurs d’hypoglycémie ont été rapportés chez 4 (0,2 %) patients de chacun des groupes dapagliflozine et placebo. Dans l’étude DELIVER, des événements majeurs d’hypoglycémie ont été rapportés chez 6 (0,2 %) patients dans le groupe dapagliflozine et 7 (0,2 %) dans le groupe placebo. Ces événements majeurs d’hypoglycémie n’ont été observés que chez les patients atteints de diabète de type 2.
Dans l’étude DAPA-CKD, des événements majeurs d’hypoglycémie ont été rapportés chez 14 (0,7 %) patients dans le groupe dapagliflozine et chez 28 (1,3 %) patients dans le groupe placebo. Ils ont été observés uniquement chez les patients atteints de diabète de type 2.
Déplétion volémique
Dans l’analyse poolée de 13 études visant à analyser la tolérance, des effets évocateurs d’une déplétion volémique (y compris, des cas de déshydratation, d’hypovolémie ou d’hypotension) ont été rapportés chez 1,1 % et 0,7 % des patients ayant reçu respectivement la dapagliflozine 10 mg et le placebo. Des réactions graves sont survenues chez < 0,2 % des patients, et se sont réparties de manière équilibrée entre les patients traités par dapagliflozine 10 mg et le placebo (voir rubrique 4.4).
Dans l’étude DECLARE, les nombres de patients présentant des événements évocateurs d’une déplétion volémique étaient équilibrés entre les groupes de traitement : 213 (2,5 %) et 207 (2,4 %) respectivement, dans les groupes dapagliflozine et placebo. Des événements indésirables graves ont été rapportés chez 81 (0,9 %) et 70 (0,8 %) des patients, dans les groupes dapagliflozine et placebo, respectivement. Les événements étaient globalement équilibrés entre les groupes de traitement dans les sous-groupes constitués en fonction de l’âge, de l’utilisation de diurétiques, de la pression artérielle et de l’utilisation d’inhibiteurs de l’enzyme de conversion (IEC)/ antagonistes des récepteurs de l'angiotensine II (ARA-II). Chez les patients présentant un DFGe <60 mL/min/1,73 m2 à l’inclusion, il y a eu 19 événements indésirables graves évocateurs d’une déplétion volémique dans le groupe dapagliflozine et 13 événements dans le groupe placebo.
Dans l’étude DAPA-HF, le nombre de patients présentant des événements indésirables évocateurs d’une déplétion volémique était de 170 (7,2 %) patients dans le groupe dapagliflozine et 153 (6,5 %) dans le groupe placebo. Moins de patients ont présenté des événements indésirables graves évocateurs d’une déplétion volémique dans le groupe dapagliflozine par rapport au groupe placebo : 23 (1,0 %) et 38 (1,6 %) patients, respectivement. Des résultats similaires ont été observés indépendamment de la présence ou non d’un diabète à l’inclusion et des valeurs initiales du DFGe. Dans l’étude DELIVER, le nombre de patients présentant des événements graves évocateurs d’une déplétion volémique était de 35 (1,1 %) dans le groupe dapagliflozine et de 31 (1,0 %) dans le groupe placebo.
Dans l’étude DAPA-CKD, le nombre de patients présentant des événements indésirables évocateurs d’une déplétion volémique était de 120 (5,6 %) dans le groupe dapagliflozine et de 84 (3,9 %) dans le groupe placebo. Il y a eu 16 (0,7 %) patients avec des événements graves à type de symptômes évocateurs d’une déplétion volémique dans le groupe dapagliflozine et 15 (0,7 %) patients dans le groupe placebo.
Acidocétose diabétique dans le diabètes de type 2
Dans l’étude DECLARE, avec une durée d’exposition médiane de 48 mois, des événements de type ACD ont été rapportés chez 27 patients du groupe dapagliflozine 10 mg et chez 12 patients du groupe placebo. Les événements sont survenus de manière homogène tout au long de la période d’étude. Sur les 27 patients ayant présenté des événements de type ACD dans le groupe dapagliflozine, 22 recevaient également un traitement par insuline au moment de l’événement. Les facteurs déclenchants de l’ACD étaient ceux attendus pour une population de patients atteints de diabète de type 2 (voir rubrique 4.4).
Dans l’étude DAPA-HF, des événements de type acidocétose diabétique (ACD) ont été rapportés chez 3 patients atteints de diabète de type 2 dans le groupe dapagliflozine et aucun dans le groupe placebo. Dans l’étude DELIVER, des événements de type ACD ont été rapportés chez 2 patients atteints de diabète de type 2 dans le groupe dapagliflozine et aucun dans le groupe placebo.
Dans l’étude DAPA-CKD, des événements de type ACD n’ont été rapportés chez aucun patient dans le groupe dapagliflozine et chez 2 patients atteints de diabète de type 2 dans le groupe placebo.
Infections des voies urinaires
Dans l’analyse poolée de 13 études visant à analyser la tolérance, les infections des voies urinaires ont été plus fréquemment rapportées chez les patients ayant reçu dapagliflozine 10 mg comparativement au placebo (respectivement, 4,7 % versus 3,5 % ; voir rubrique 4.4). La plupart des infections étaient légères à modérées, les patients ont répondu à un traitement standard initial et ont rarement entraîné l’arrêt du traitement par dapagliflozine. Ces infections ont été plus fréquentes chez les femmes, et les patients ayant un antécédent étaient plus susceptibles d’avoir une infection récurrente.
Dans l’étude DECLARE, les événements graves de type infections des voies urinaires ont été rapportés moins fréquemment avec la dapagliflozine 10 mg par rapport au placebo, à savoir 79 (0,9 %) événements versus 109 (1,3 %) événements, respectivement.
Dans l’étude DAPA-HF, le nombre de patients présentant des événements indésirables graves de type infections des voies urinaires était de 14 (0,6 %) patients dans le groupe dapagliflozine et 17 (0,7 %) dans le groupe placebo. Cinq (0,2 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections des voies urinaires dans chacun des groupes dapagliflozine et placebo. Dans l’étude DELIVER, le nombre de patients présentant des événements indésirables graves de type infections des voies urinaires était de 41 (1,3 %) dans le groupe dapagliflozine et de 37 (1,2 %) dans le groupe placebo. Treize (0,4 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections des voies urinaires dans le groupe dapagliflozine et 9 (0,3 %) dans le groupe placebo.
Dans l’étude DAPA-CKD, le nombre de patients présentant des événements indésirables graves de type infections des voies urinaires était de 29 (1,3 %) dans le groupe dapagliflozine et de 18 (0,8 %) dans le groupe placebo. Huit (0,4 %) patients ont présenté des événements indésirables entraînant l’arrêt du traitement en raison d’infections des voies urinaires dans le groupe dapagliflozine et 3 (0,1 %) dans le groupe placebo. Le nombre de patients rapportant des événements indésirables graves ou des arrêts du traitement en raison d’événements indésirables de type infections des voies urinaires parmi les patients non diabétiques était similaire entre les groupes de traitement (6 [0,9 %] versus 4 [0,6 %] pour les événements indésirables graves, et 1 [0,1 %] versus 0 pour les arrêts du traitement en raison d’événements indésirables, dans le groupe dapagliflozine et le groupe placebo, respectivement).
Augmentation de la créatinine
Les effets indésirables liés à une augmentation de la créatinine ont été regroupés (par ex : diminution de la clairance de la créatinine rénale, altération de la fonction rénale, augmentation de la créatininémie et diminution du débit de filtration glomérulaire). Dans l’analyse poolée de 13 études visant à analyser la tolérance, ce groupe d’effets indésirables a été rapporté respectivement chez 3,2 % des patients recevant la dapagliflozine 10 mg et chez 1,8 % des patients recevant le placebo. Chez les patients avec une fonction rénale normale ou une altération légère de la fonction rénale (valeur initiale du DFGe ≥ 60 mL/min/1,73 m2), ce groupe d’effets indésirables a été rapporté chez 1,3 % des patients recevant la dapagliflozine 10 mg et chez 0,8 % des patients recevant le placebo. Ces réactions ont été plus fréquentes chez les patients avec une valeur initiale du DFGe ≥ 30 et <60 mL/min/1,73 m2 (18,5 % dapagliflozine 10 mg versus 9,3 % placebo).
Des évaluations complémentaires des patients qui avaient présenté des événements indésirables liés à un trouble rénal ont montré que la plupart des patients avaient des modifications de la créatininémie inférieures ou égales à 44 micromoles/L (0,5 mg/dL) par rapport à la valeur initiale. Les augmentations de la créatinine ont été généralement transitoires lors d’un traitement continu ou réversibles après l’arrêt du traitement.
Dans l’étude DECLARE, incluant des patients âgés et des patients présentant une insuffisance rénale (DFGe inférieur à 60 mL/min/1,73 m2), le DFGe a diminué avec le temps dans les deux groupes de traitement. À 1 an, le DFGe moyen était légèrement plus faible, et à 4 ans, le DFGe moyen était légèrement plus élevé dans le groupe dapagliflozine que dans le groupe placebo.
Dans les études DAPA-HF et DELIVER, le DFGe a diminué au fil du temps dans le groupe dapagliflozine et le groupe placebo. Dans l’étude DAPA-HF, la diminution initiale du DFGe moyen était de -4,3 mL/min/1,73 m2 dans le groupe dapagliflozine et de -1,1 mL/min/1,73 m2 dans le groupe placebo. À 20 mois, la variation par rapport à la valeur initiale du DFGe était similaire entre les groupes de traitement : -5,3 mL/min/1,73 m2 pour la dapagliflozine et -4,5 mL/min/1,73 m2 pour le placebo. Dans l’étude DELIVER, la diminution du DFGe moyen à un mois était de ‑3,7 mL/min/1,73 m2 dans le groupe dapagliflozine et de ‑0,4 mL/min/1,73 m2 dans le groupe placebo. À 24 mois, la variation par rapport à la valeur initiale du DFGe était similaire entre les groupes de traitement : ‑4,2 mL/min/1,73 m2 dans le groupe dapagliflozine et ‑3,2 mL/min/1,73 m2 dans le groupe placebo.
Dans l’étude DAPA-CKD, le DFGe a diminué au fil du temps dans le groupe dapagliflozine et le groupe placebo. La diminution initiale (J1) du DFGe moyen était de -4,0 mL/min/1,73 m2 dans le groupe dapagliflozine et de -0,8 mL/min/1,73 m2 dans le groupe placebo. À 28 mois, la variation par rapport à la valeur initiale du DFGe était de -7,4 mL/min/1,73 m2 dans le groupe dapagliflozine et de ‑8,6 mL/min/1,73 m2 dans le groupe placebo.
Population pédiatrique
Le profil de sécurité de la dapagliflozine observé dans une étude clinique chez des enfants âgés de 10 ans et plus atteints d’un diabète de type 2 (voir rubrique 5.1) était cohérent avec celui observé dans les études chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance :
Site internet : www.notifieruneffetindesirable.be
e-mail : adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
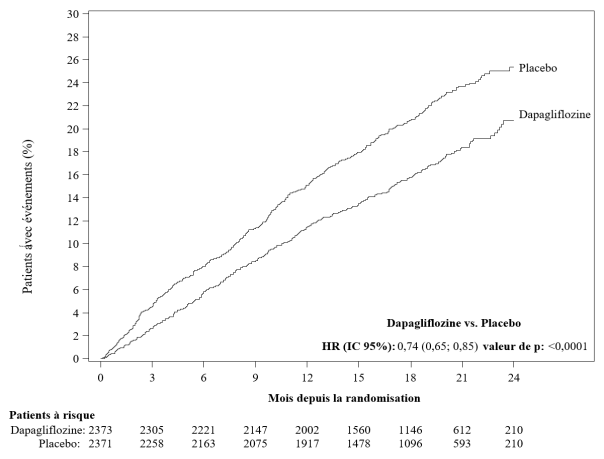
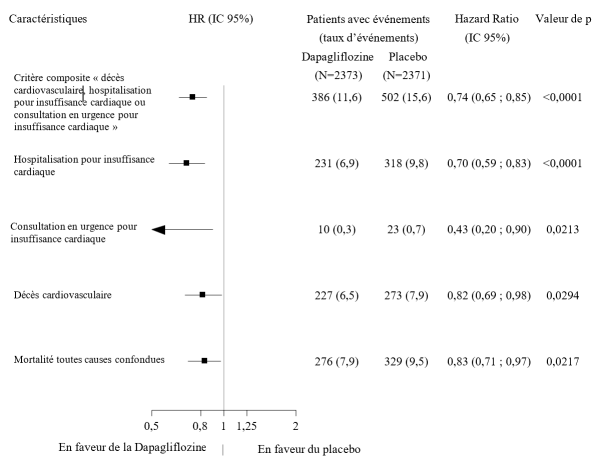
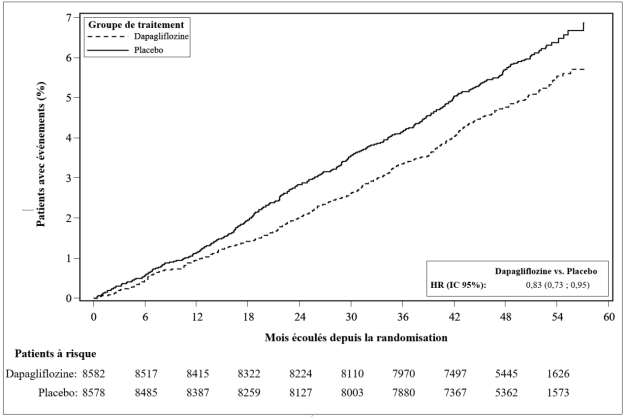 « Patients à risque » correspond au nombre de patients à risque au début de la période.
« Patients à risque » correspond au nombre de patients à risque au début de la période. 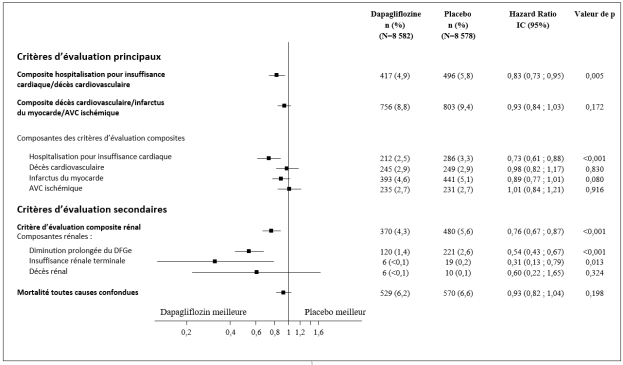
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
AstraZeneca AB
SE‑151 85 Södertälje
Suède
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
Forxiga 5 mg, comprimés pelliculés
EU/1/12/795/001 14 comprimés pelliculés
EU/1/12/795/002 28 comprimés pelliculés
EU/1/12/795/003 98 comprimés pelliculés
EU/1/12/795/004 30 x 1 (unidose) comprimés pelliculés
EU/1/12/795/005 90 x 1 (unidose) comprimés pelliculés
Forxiga 10 mg, comprimés pelliculés
EU/1/12/795/006 14 comprimés pelliculés
EU/1/12/795/007 28 comprimés pelliculés
EU/1/12/795/008 98 comprimés pelliculés
EU/1/12/795/009 30 x 1 (unidose) comprimés pelliculés
EU/1/12/795/010 90 x 1 (unidose) comprimés pelliculés
EU/1/12/795/011 10 x 1 (unidose) comprimés pelliculés
10. DATE DE MISE A JOUR DU TEXTE
03/2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3018165 | FORXIGA 10 MG COMP PELL 98 X 10 MG | A10BK01 | € 144,17 | - | Oui | € 15,9 | € 10,5 |
| 3018173 | FORXIGA 10 MG COMP PELL 28 X 10 MG | A10BK01 | € 47,7 | - | Oui | € 2 | € 1 |