ANNEXE I
RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Praluent 75 mg, solution injectable en stylo pré-rempli
Praluent 150 mg, solution injectable en stylo pré-rempli
Praluent 75 mg, solution injectable en seringue pré-remplie
Praluent 150 mg, solution injectable en seringue pré-remplie
Praluent 300 mg, solution injectable en stylo pré-rempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Praluent 75 mg, solution injectable en stylo pré-rempli
Chaque stylo pré-rempli à usage unique contient 75 mg d’alirocumab dans 1 ml de solution.
Praluent 75 mg, solution injectable en seringue pré-remplie
Chaque seringue pré-remplie à usage unique contient 75 mg d’alirocumab dans 1 ml de solution.
Praluent 150 mg, solution injectable en stylo pré-rempli
Chaque stylo pré-rempli à usage unique contient 150 mg d’alirocumab dans 1 ml de solution.
Praluent 300 mg, solution injectable en stylo pré-rempli
Chaque stylo pré-rempli à usage unique contient 300 mg d’alirocumab dans 2 ml de solution.
Praluent 150 mg, solution injectable en seringue pré-remplie
Chaque seringue pré-remplie à usage unique contient 150 mg d’alirocumab dans 1 ml de solution.
Alirocumab est un anticorps monoclonal humain de type Immunoglobuline G1 (IgG1) produit dans des cellules ovariennes de hamster chinois par la technique de l’ADN recombinant.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable (injection) en stylo pré-rempli/en seringue pré-remplie.
Solution limpide, incolore à jaune pâle.
pH :5,7 - 6,3
Osmolarité :
Praluent 75 mg solution injectable
293 – 439mOsm/kg
Praluent 150 mg solution injectable
383 – 434mOsm/kg
Praluent 300 mg solution injectable
383 – 434mOsm/kg
4. DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Hypercholestérolémie primaire et dyslipidémie mixte
Praluent est indiqué chez l’adulte présentant une hypercholestérolémie primaire (hétérozygote familiale et non familiale) ou une dyslipidémie mixte, et chez les enfants à partir de 8 ans présentant une hypercholestérolémie familiale hétérozygote (HFHe) en complément d’un régime alimentaire :
en association avec une statine seule ou une statine avec d’autres thérapies hypolipémiantes chez les patients ne pouvant atteindre leur objectif de LDL-C, sous statine à dose maximale tolérée ou,
- seul ou en association avec d’autres thérapies hypolipémiantes chez les patients intolérants aux statines, ou chez qui les statines sont contre-indiquées.
Maladie cardiovasculaire athéroscléreuse établie
Praluent est indiqué chez les adultes avec une maladie cardiovasculaire athéroscléreuse établie pour réduire le risque cardiovasculaire en diminuant les taux de LDL-C, en complément de la correction des autres facteurs de risque :
- en association avec une statine à la dose maximale tolérée avec ou sans autres thérapies hypolipémiantes ou,
- seul ou en association avec d’autres thérapies hypolipémiantes chez les patients intolérants aux statines, ou chez qui les statines sont contre-indiquées.
Pour les résultats des études concernant les effets sur le LDL-C, les événements cardiovasculaires et les populations étudiées, voir rubrique 5.1.
4.2 Posologie et mode d’administration
Posologie
Adultes
Avant de débuter un traitement par alirocumab, toute cause secondaire d’hypercholestérolémie ou de dyslipidémie mixte doit être éliminée (par ex. syndrome néphrotique, hypothyroïdie).
Les doses recommandées d’alirocumab sont de 75 mg une fois toutes les 2 semaines, 150 mg une fois toutes les 2 semaines, 300 mg une fois toutes les 4 semaines (mensuellement), administrées par voie sous-cutanée. Toutes les doses peuvent être utilisées pour initier le traitement.
La dose d’alirocumab peut être ajustée individuellement en fonction des caractéristiques du patient telles que son taux de LDL-C avant traitement, son objectif thérapeutique et sa réponse au traitement. Les paramètres lipidiques peuvent être évalués 4 à 8 semaines après l’instauration ou l’ajustement posologique du traitement, la posologie pouvant alors être ajustée en fonction des résultats (augmentation ou diminution de la dose le cas échéant). Une réduction importante du taux de LDL-C est attendue avec l’alirocumab 150 mg une fois toutes les 2 semaines et 300 mg une fois toutes les 4 semaines (mensuellement), la posologie maximale étant 150 mg une fois toutes les 2 semaines (voir rubrique 5.1).
Hypercholestérolémie familiale hétérozygote chez les patients pédiatriques à partir de 8 ans
Poids corporel des patients | Dose recommandée | Dose recommandée si une réduction supplémentaire du LDL-C est nécessaire* |
Moins de 50 kg | 150 mg une fois toutes les 4 semaines | 75 mg une fois toutes les 2 semaines |
50 kg ou plus | 300 mg une fois toutes les 4 semaines | 150 mg une fois toutes les 2 semaines |
*Les taux de lipides peuvent être réévalués 8 semaines après l’initiation ou la titration du traitement et la dose peut être ajustée en conséquence.
Dose oubliée
En cas d’oubli d’une dose, la dose doit être administrée dès que possible, puis le traitement doit être repris selon le schéma posologique initial.
Populations particulières
Personnes âgées
Aucun ajustement posologique n’est nécessaire chez les patients âgés.
Insuffisance hépatique
Aucun ajustement posologique n’est nécessaire chez les patients atteints d’insuffisance hépatique légère à modérée. Aucune donnée n’est disponible pour les patients atteints d’insuffisance hépatique sévère (voir rubrique 5.2).
Insuffisance rénale
Aucun ajustement posologique n’est nécessaire chez les patients atteints d’insuffisance rénale légère à modérée. Peu de données sont disponibles chez les patients atteints d’insuffisance rénale sévère (voir rubrique 5.2).
Poids corporel
Aucun ajustement posologique n’est nécessaire en fonction du poids des patients.
Population pédiatrique
La sécurité et l’efficacité de Praluent chez les enfants âgés de moins de 8 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie sous-cutanée.
L’alirocumab est administré par injection sous-cutanée dans la cuisse, l’abdomen ou le haut du bras.
Chaque stylo pré-rempli ou seringue pré-remplie est à usage unique exclusivement.
Pour administrer la dose de 300 mg, une injection de 300 mg, ou deux injections de 150 mg consécutives sur deux sites d’injection différents doivent être effectuées.
Il est recommandé d’alterner les sites d’injection à chaque injection.
L’alirocumab ne doit pas être injecté dans des zones d’affections cutanées actives ou dans des zones lésées, telles qu’érythèmes solaires, éruptions cutanées, zones inflammatoires ou infectées.
L’alirocumab ne doit pas être administré en même temps que d’autres médicaments injectables au même site d’injection.
Précautions à prendre avant la manipulation ou l’administration
La solution doit être amené à température ambiante avant l’utilisation (voir rubrique 6.6).
Population pédiatrique à partir de 8 ans
Chez les adolescents à partir de 12 ans, il est recommandé d’administrer Praluent par ou sous la supervision d’un adulte.
Chez les enfants de moins de 12 ans, Praluent doit être administré par un soignant ou un accompagnant.
Adultes
Après une formation sur la bonne technique d’injection sous-cutanée par un professionnel de santé, le patient adulte peut s’injecter lui-même l’alirocumab ou un soignant ou un accompagnant peut le lui administrer.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquents, aux doses recommandées, sont des réactions locales au site d’injection (6,1%), des symptômes des voies aériennes supérieures (2,0%) et un prurit (1,1%). Les effets indésirables les plus fréquents conduisant à un arrêt du traitement chez les patients traités par alirocumab étaient des réactions locales au site d’injection.
Le profil de sécurité dans l’étude ODYSSEY OUTCOMES était conforme au profil global de sécurité décrit dans les essais contrôlés de Phase 3.
Aucune différence n’a été observée dans le profil de sécurité entre les deux doses (75 mg et 150 mg) utilisées durant le programme de Phase 3.
Tableau résumé des effets indésirables
Les effets indésirables suivants ont été rapportés chez les patients traités par l’alirocumab dans les études contrôlées poolées et/ou l’utilisation après commercialisation (voir tableau 1).
Les fréquences de tous les effets indésirables identifiés au cours des essais cliniques ont été calculées en fonction de leur incidence dans les essais cliniques de Phase 3 poolés. Les effets indésirables sont présentés selon le système de classification par organe. Les catégories de fréquence sont définies de la manière suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
La fréquence des effets indésirables rapportés au cours de l’utilisation après commercialisation ne peut pas être déterminée car ils proviennent de déclarations spontanées. En conséquence, la fréquence de ces effets indésirables est qualifiée d’« indéterminée ».
Tableau 1. Effets indésirables
Classe de système d’organes | Fréquent | Rare | Fréquence |
Affections du système immunitaire |
| Hypersensibilité, |
|
Affections respiratoires, thoraciques et médiastinales | Symptômes des voies aériennes supérieures* |
|
|
Affections de la peau et du tissu sous-cutané | Prurit | Urticaire, | Angioedème |
Troubles généraux et anomalies au site d’administration | Réactions au site d’injection** |
| Syndrome pseudo-grippal |
* incluant principalement douleurs oropharyngées, rhinorrhées, éternuements.
** incluant érythème/rougeur, démangeaisons, gonflement, douleur/sensibilité.
Description de certains effets indésirables
Réactions locales au site d’injection
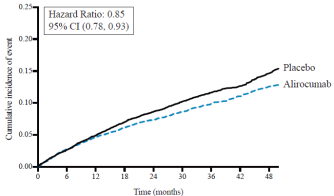
Des réactions locales au site d’injection, notamment érythème/rougeur, démangeaisons, gonflement, douleur/sensibilité ont été rapportées chez 6,1 % des patients traités par l’alirocumab versus 4,1 % dans le groupe contrôle (recevant des injections de placebo). La plupart des réactions au site d’injection étaient transitoires et de faible intensité. Les taux d’arrêt de traitement dû à des réactions locales au site d’injection étaient comparables entre les deux groupes (0,2 % dans le groupe alirocumab versus 0,3 % dans le groupe contrôle). Dans l’étude de morbimortalité cardiovasculaire (ODYSSEY OUTCOMES), les réactions au site d’injection sont aussi survenues plus fréquemment chez les patients traités par alirocumab que chez les patients traités par placebo (3,8 % sous alirocumab versus 2,1 % sous placebo).
Réactions allergiques générales
Des réactions allergiques générales ont été rapportées plus fréquemment dans le groupe alirocumab (8,1 % des patients) que dans le groupe contrôle (7,0 % des patients) principalement en raison d’une différence dans l’incidence du prurit. Les cas de prurit observés étaient généralement légers et transitoires. De plus, des réactions allergiques rares et parfois graves, telles que l’hypersensibilité, l’eczéma nummulaire, l’urticaire et la vascularite d’hypersensibilité ont été rapportées dans les études cliniques contrôlées (voir rubrique 4.4). Dans l’étude de morbimortalité cardiovasculaire (ODYSSEY OUTCOMES), les réactions allergiques générales étaient similaires chez les patients traités par alirocumab et chez les patients traités par placebo (7,9 % sous alirocumab, 7,8 % sous placebo). Aucune différence n’a été observée dans l’incidence du prurit.
Populations particulières
Personnes âgées
Bien qu’aucun problème de sécurité n’ait été observé chez les patients âgés de plus de 75 ans, les données sont limitées dans ce groupe d’âge.
Dans les études contrôlées de Phase 3 portant sur l’hypercholestérolémie primaire et la dyslipidémie mixte, 1 158 patients (34,7 %) traités par alirocumab étaient âgés de ≥ 65 ans, et 241 patients (7,2 %) traités par alirocumab étaient âgés de ≥ 75 ans. Dans l’étude contrôlée de morbimortalité cardiovasculaire, 2 505 patients (26,5 %) traités par alirocumab étaient âgés de ≥ 65 ans, et 493 patients (5,2 %) traités par alirocumab étaient âgés de ≥ 75 ans. Aucune différence significative quant à la sécurité ou l’efficacité n’a été observée avec l’augmentation de l’âge.
Population pédiatrique
La sécurité et l’efficacité de Praluent ont été établies chez les enfants et les adolescents atteints d’hypercholestérolémie familiale hétérozygote (HFHe). Une étude clinique visant à évaluer les effets de Praluent a été menée chez 153 patients âgés de 8 à 17 ans atteints de HFHe. Aucun nouveau résultat en matière de sécurité n’a été identifié et les données de sécurité dans cette population étaient cohérentes avec le profil de sécurité connu du produit chez les adultes atteints de HFHe.
L’expérience relative à l’alirocumab chez les patients pédiatriques atteints d’hypercholestérolémie familiale homozygote (HFHo) est limitée à 18 patients âgés de 8 à 17 ans. Aucun nouveau constat de sécurité n’a été observé par rapport au profil de sécurité connu chez l’adulte.
Étude du schéma posologique avec une fréquence d’administration d’une fois toutes les 4 semaines
Le profil de tolérance chez les patients traités avec 300 mg toutes les 4 semaines (mensuellement) était similaire au profil de tolérance décrit dans le programme d’études cliniques utilisant un schéma posologique avec une fréquence d’administration d’une fois toutes les 2 semaines, excepté pour un taux plus élevé de réactions locales au site d’injection. Les réactions locales au site d’injection ont été rapportées chez 16,6% des patients du groupe recevant alirocumab 300 mg toutes les 4 semaines et 7,9% dans le groupe contrôle. Les patients du groupe recevant alirocumab 300 mg toutes les 4 semaines recevaient alternativement des injections de placebo pour ne pas lever l’insu en regard de la fréquence des injections. En dehors des réactions locales au site d’injection (ISRs) survenant après injection de placebo, la fréquence des ISRs était de 11.8%. Le taux de sortie d’étude suite aux réactions au site d’injection a été de 0,7% dans le groupe 300 mg toutes les 4 semaines versus 0% dans le groupe contrôle.
Valeurs de LDL-C < 0,65 mmol/L (< 25 mg/dL)
Dans l’ensemble des études cliniques les traitements hypolipémiants de fond n’ont pas pu être ajustés du fait du design des études. Le pourcentage de patients ayant atteint des valeurs de LDL-C < 0,65 mmol/L (< 25 mg/dL) dépendait à la fois du taux initial de LDL-C et de la dose d’alirocumab.
Dans un pool d’études contrôlées avec la dose initiale de 75 mg toutes les 2 semaines (Q2S) et augmentée à 150 mg Q2S si le taux de LDL-C du patient n’était pas < 1,81 mmol/L ou < 2,59 mmol/L (< 70 mg/dL ou < 100 mg/dL) : 29,3 % des patients dont le taux initial de LDL-C était < 2,59 mmol/L (< 100 mg/dL) et 5,0 % des patients dont le taux initial de LDL-C était ≥ 2,59 mmol/L (≥ 100 mg/dL), traités par alirocumab ont présenté deux valeurs consécutives de LDL-C < 0,65 mmol/L (< 25 mg/dL). Dans l’étude ODYSSEY OUTCOMES la dose initiale d’alirocumab était de 75 mg Q2S et était augmentée à 150 mg Q2S si le taux de LDL-C du patient n’était pas < 1,29 mmol/L (< 50 mg/dL) : 54,8 % des patients dont le taux initial de LDL-C était < 2,59 mmol/L (< 100 mg/dL) et 24,2 % des patients dont le taux initial de LDL-C était ≥ 2,59 mmol/L (≥ 100 mg/dL), traités par alirocumab ont présenté deux valeurs consécutives de LDL-C < 0,65 mmol/L (< 25 mg/dL).
Bien qu’aucune conséquence défavorable de très faibles taux de LDL-C n’ait été identifiée dans le cadre des essais avec l’alirocumab, les effets à long terme de taux très bas de LDL-C prolongés dans le temps sont inconnus.
Immunogénicité / Anticorps anti-médicament (Anti-drug-antibodies (ADA))
Dans l’étude ODYSSEY OUTCOMES, 5,5 % des patients traités par alirocumab 75 mg et/ou 150 mg toutes les 2 semaines (Q2S) ont eu des anticorps anti-médicament (ADA) détectés après le début du traitement versus 1,6 % des patients traités par placebo, la plupart étaient transitoires. Des réponses ADA persistantes ont été observées chez 0,7 % des patients traités par alirocumab et chez 0,4 % des patients traités par placebo. Des anticorps neutralisants (AcN) ont été observées chez 0,5 % des patients traités par alirocumab et chez < 0,1 % des patients traités par placebo.
Les réponses ADA, y compris les anticorps neutralisants, avaient un titre faible et n’ont pas semblé avoir un impact cliniquement significatif sur l’efficacité ou la sécurité de l’alirocumab, à l’exception d’un taux plus élevé de réactions au site d’injection chez les patients ayant présenté des ADA sous traitement, comparativement aux patients n’ayant pas présenté d’ADA (7,5 % versus 3,6 %). Les conséquences à long terme de la poursuite du traitement par alirocumab en présence d’ADA sont inconnues.
Dans un groupe de dix essais contrôlés versus placebo et versus substance active dans lesquels les patients étaient traités par alirocumab 75 mg et/ou 150 mg Q2S, ainsi que dans une étude clinique distincte dans laquelle les patients étaient traités par alirocumab 75 mg Q2S ou 300 mg toutes les 4 semaines (incluant certains patients dont la dose a été ajustée à 150 mg Q2S), l’incidence de détection d’ADA et d’AcN était similaires aux résultats de l’essai ODYSSEY OUTCOMES décrit ci-dessus.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique : Agence Fédérale des Médicaments et des Produits de Santé : www.afmps.be – Division Vigilance : Site internet : www.notifieruneffetindesirable.be – E-mail : adr@fagg-afmps.be
Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé – Site internet : www.guichet.lu/pharmacovigilance.

7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Sanofi Winthrop Industrie
82 avenue Raspail
94250 Gentilly
France
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/15/1031/001
EU/1/15/1031/002
EU/1/15/1031/003
EU/1/15/1031/004
EU/1/15/1031/005
EU/1/15/1031/006
EU/1/15/1031/007
EU/1/15/1031/008
EU/1/15/1031/009
EU/1/15/1031/010
EU/1/15/1031/011
EU/1/15/1031/012
EU/1/15/1031/013
EU/1/15/1031/014
EU/1/15/1031/015
EU/1/15/1031/016
EU/1/15/1031/017
EU/1/15/1031/018
EU/1/15/1031/019
EU/1/15/1031/020
EU/1/15/1031/021
EU/1/15/1031/022
EU/1/15/1031/023
EU/1/15/1031/024
10. DATE DE MISE À JOUR DU TEXTE
10/2025
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’Agence européenne des médicaments http://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3365103 | PRALUENT 75MG SOL INJ STYLO PREREMPLI 2 X 75MG | C10AX14 | € 605,79 | - | Oui | € 2 | € 1 |
| 3365111 | PRALUENT 150MG SOL INJ STYLO PREREMPLI 2 X 150MG | C10AX14 | € 605,79 | - | Oui | € 2 | € 1 |
| 3365129 | PRALUENT 75MG SOL INJ STYLO PREREMPLI 6 X 75MG | C10AX14 | € 1438,53 | - | Oui | € 2 | € 1 |
| 3365137 | PRALUENT 150MG SOL INJ STYLO PREREMPLI 6 X 150MG | C10AX14 | € 1438,53 | - | Oui | € 2 | € 1 |
| 4565875 | PRALUENT 300MG SOL INJ STYLO PREREMPLI 3 X 300MG | € 1438,53 | - | Oui | € 2 | € 1 |