RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Trulicity 0,75 mg solution injectable en stylo pré‑rempli
Trulicity 1,5 mg solution injectable en stylo pré‑rempli
Trulicity 3 mg solution injectable en stylo pré‑rempli
Trulicity 4,5 mg solution injectable en stylo pré‑rempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Trulicity 0,75 mg solution injectable en stylo pré‑rempli
Chaque stylo pré‑rempli contient 0,75 mg de dulaglutide* dans 0,5 mL de solution.
Excipient à effet notoire :
Un mL de solution contient 0,20 mg de polysorbate 80.
Trulicity 1,5 mg solution injectable en stylo pré‑rempli
Chaque stylo pré‑rempli contient 1,5 mg de dulaglutide* dans 0,5 mL de solution.
Excipient à effet notoire :
Un mL de solution contient 0,20 mg de polysorbate 80.
Trulicity 3 mg solution injectable en stylo pré‑rempli
Chaque stylo pré‑rempli contient 3 mg de dulaglutide* dans 0,5 mL de solution.
Excipient à effet notoire :
Un mL de solution contient 0,25 mg de polysorbate 80.
Trulicity 4,5 mg solution injectable en stylo pré‑rempli
Chaque stylo pré‑rempli contient 4,5 mg de dulaglutide* dans 0,5 mL de solution.
Excipient à effet notoire :
Un mL de solution contient 0,25 mg de polysorbate 80.
*produit sur cellules CHO par la technique d'ADN recombinant.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable.
Solution limpide, incolore.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Diabète de type 2
Trulicity est indiqué chez les patients de 10 ans et plus pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d’un régime alimentaire et d’une activité physique :
- en monothérapie, quand l’utilisation de la metformine est considérée comme inappropriée en raison d'une intolérance ou de contre-indications.
- en association avec d'autres médicaments destinés au traitement du diabète.
Pour les résultats des études concernant les associations, les effets sur le contrôle glycémique et les évènements cardiovasculaires, ainsi que sur les populations étudiées, voir les rubriques 4.4, 4.5 et 5.1.
4.2 Posologie et mode d'administration
Posologie
Adultes
En monothérapie
La dose recommandée est de 0,75 mg une fois par semaine.
En association
La dose recommandée est de 1,5 mg une fois par semaine.
Si nécessaire,
- la dose de 1,5 mg peut être augmentée, après 4 semaines au moins, à 3 mg une fois par semaine ;
- la dose de 3 mg peut être augmentée, après 4 semaines au moins, à 4,5 mg une fois par semaine.
La dose maximale est de 4,5 mg une fois par semaine.
Population pédiatrique
La dose initiale pour les patients pédiatriques âgés de 10 ans et plus est de 0,75 mg une fois par semaine.
Si nécessaire, la dose peut être augmentée à 1,5 mg une fois par semaine après 4 semaines au moins. La dose maximale est de 1,5 mg une fois par semaine.
En association
Lorsque Trulicity est ajouté à un traitement en cours par metformine et/ou pioglitazone, la dose de metformine et/ou de pioglitazone peut être conservée. Lorsque Trulicity est ajouté à un traitement en cours par metformine et/ou inhibiteur du co-transporteur de sodium-glucose de type 2 (iSGLT2), la dose de metformine et/ou de l’iSGLT2 peut être conservée. Lorsqu’il est ajouté à un traitement en cours par sulfamide hypoglycémiant ou insuline, une diminution de la dose de sulfamide hypoglycémiant ou d'insuline peut être envisagée afin de réduire le risque d'hypoglycémie (voir rubriques 4.4 et 4.8).
L'utilisation de Trulicity ne nécessite pas d'auto‑surveillance glycémique. Une auto-surveillance glycémique est nécessaire pour ajuster la dose de sulfamide hypoglycémiant ou d’insuline, notamment lors de l’instauration du traitement par Trulicity et de la réduction des doses d’insuline. L’adoption d’une approche par étapes de la réduction des doses d’insuline est recommandée.
Doses oubliées
En cas d'oubli, la dose doit être administrée le plus rapidement possible si le délai avant la date de la prochaine dose est d'au moins 3 jours (72 heures). Si la dose suivante est prévue dans moins de 3 jours (72 heures), la dose oubliée ne doit pas être administrée et la dose suivante doit être administrée le jour normalement prévu. Dans tous les cas, les patients peuvent ensuite reprendre leur schéma d’administration hebdomadaire habituel.
Populations particulières
Patients âgés
Aucun ajustement de la dose n'est requis en fonction de l'âge (voir rubrique 5.2).
Insuffisants rénaux
Aucun ajustement de la dose n'est requis chez les patients atteints d'insuffisance rénale légère, modérée ou sévère (DFGe < 90 à ≥ 15 mL/min/1,73m2).
L'expérience chez les patients présentant une insuffisance rénale terminale (< 15 mL/min/1,73m2) étant très limitée, Trulicity ne peut pas être recommandé chez ces patients (voir rubriques 5.1 et 5.2).
Insuffisants hépatiques
Aucun ajustement de la dose n'est requis chez les patients atteints d'insuffisance hépatique.
Population pédiatrique
La sécurité et l'efficacité du dulaglutide chez les enfants de moins de 10 ans n'ont pas été établies et aucune donnée n'est disponible (voir rubriques 5.1 et 5.2).
Mode d'administration
Trulicity doit être administré par injection sous-cutanée dans l'abdomen, la cuisse ou le haut du bras. Il ne doit pas être administré par injection intraveineuse ou intramusculaire.
La dose peut être administrée à toute heure de la journée, au moment ou en dehors des repas.
Le jour de la semaine prévu pour l'injection peut être éventuellement modifié, dans la mesure où la dernière dose a été administrée au moins 3 jours (72 heures) avant.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
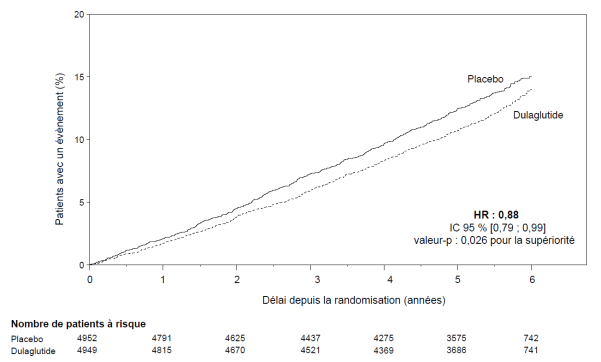
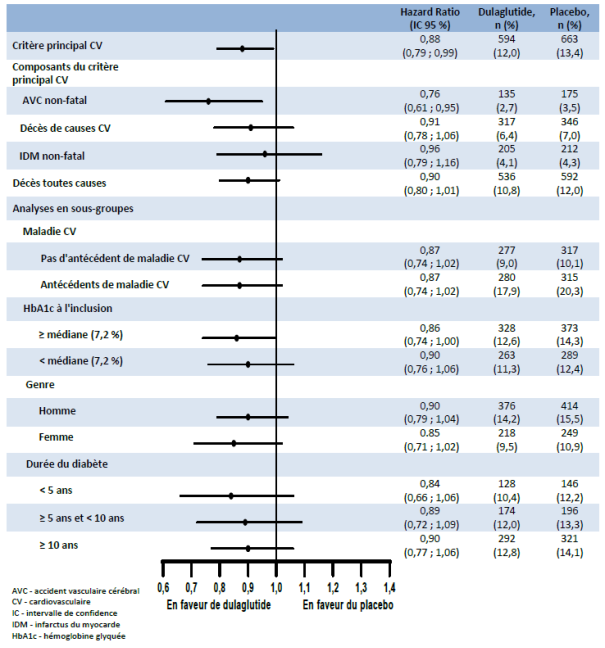
Au cours des essais cliniques de phases 2 et 3 réalisés pour l’enregistrement initial du dulaglutide 0,75 mg et 1,5 mg, 4 006 patients ont été exposés au dulaglutide seul ou en association avec d'autres agents hypoglycémiants. Les effets indésirables les plus fréquemment rapportés dans les essais cliniques ont été de nature gastro-intestinale, incluant nausées, vomissements et diarrhées. En général, ces effets ont été d'intensité légère à modérée et de nature transitoire. Les résultats de l’étude d’évènements cardiovasculaires à long terme avec 4 949 patients randomisés sous dulaglutide et suivis sur une durée médiane de 5,4 ans ont été cohérents avec ces données.
Liste tabulée des effets indésirables
Les effets indésirables suivants ont été identifiés à partir de l’évaluation pendant toute la durée des études cliniques de phase 2 et de phase 3, de l’étude d’évènements cardiovasculaires à long terme et des cas rapportés après commercialisation. Les effets indésirables sont listés dans le Tableau 1 selon la terminologie MedDRA par classe de système d’organe et par ordre décroissant de fréquence (très fréquent : ≥ 1/10 ; fréquent : ≥ 1/100, < 1/10 ; peu fréquent : ≥ 1/1 000, < 1/100 ; rare : ≥ 1/10 000, < 1/1 000 ; très rare : < 1/10 000 et fréquence indéterminée : ne peut être estimée sur la base des données disponibles). Dans chaque groupe, les effets indésirables sont classés par ordre décroissant de fréquence. Les fréquences des effets ont été calculées sur la base de leur incidence dans les études d’enregistrement de phases 2 et de phase 3.
Tableau 1. Fréquence des effets indésirables du dulaglutide
Classe de système d’organe | Très fréquent | Fréquent | Peu fréquent | Rare | Indétermi-née |
Affections du système immunitaire |
|
| Hypersensibili-té | Réaction anaphylacti-que# |
|
Troubles du métabolisme et de la nutrition | Hypoglycémie* (en cas d'association avec de l'insuline, du glimépiride, de la metformine† ou de la metformine plus glimépiride) | Hypoglycémie* (en monothérapie ou en association avec metformine plus pioglitazone) | Déshydratation |
|
|
Affections du système nerveux |
|
| Dysgueusie |
|
|
Affections gastro-intestinales | Nausée, diarrhée, vomissement†, douleur abdominale† | Diminution de l'appétit, dyspepsie, constipation, flatulence, distension abdominale, reflux gastro-œsophagien, éructation |
| Pancréatite aiguë, retard de la vidange gastrique | Occlusion intestinale non mécanique |
Affections hépatobiliaires |
|
| Lithiase biliaire, |
|
|
Affections de la peau et du tissu sous-cutané |
|
|
| Angio-œdème# |
|
Troubles généraux et anomalies au site d’administration |
| Fatigue | Réactions au site d'injection$ |
|
|
Investigations |
| Tachycardie sinusale, bloc auriculo-ventriculaire (BAV) de 1er degré |
|
|
|
# Rapportés après commercialisation.
* Hypoglycémie symptomatique documentée avec une glycémie ≤ 3,9 mmol/L
† Avec le dulaglutide 0,75 mg, la fréquence des effets indésirables correspond à celle du groupe de fréquence immédiatement inférieur.
$ La fréquence observée dans une étude pédiatrique était « fréquente » ; 3,9 % (2 patients) dans le groupe dulaglutide 0,75 mg, 3,8 % (2 patients) dans le groupe dulaglutide 1,5 mg et 2 % (1 patient) dans le groupe placebo. Tous les évènements étaient d’intensité légère à modérée.
Description de certains effets indésirables
Hypoglycémie
Lorsque les doses de dulaglutide de 0,75 mg et de 1,5 mg ont été utilisées en monothérapie ou en association avec de la metformine seule ou de la metformine et de la pioglitazone, les incidences d'hypoglycémie symptomatique documentée ont été de 5,9 % à 10,9 %, et les taux étaient de 0,14 à 0,62 événement/patient/an et aucun épisode d'hypoglycémie sévère n'a été rapporté.
Les incidences d'hypoglycémie symptomatique documentée avec le dulaglutide aux doses respectives de 0,75 mg et de 1,5 mg, utilisé en association avec un sulfamide hypoglycémiant et de la metformine ont été de 39,0 % et de 40,3 %, et les taux ont été de 1,67 et 1,67 événement/patient/an. Les incidences des épisodes d'hypoglycémie sévère ont été de 0 et de 0,7 % et les taux de 0,00 et de 0,01 événement/patient/an respectivement, pour chaque dose. L’incidence d'hypoglycémie symptomatique documentée avec le dulaglutide utilisé à la dose de 1,5 mg avec un sulfamide hypoglycémiant seul a été de 11,3 % ; le taux a été de 0,90 événement/patient/an, et il n’y a pas eu d’épisodes d'hypoglycémie sévère.
L’incidence d'hypoglycémie symptomatique documentée avec le dulaglutide à la dose de 1,5 mg utilisé en association avec de l’insuline glargine a été de 35,3 % et le taux a été de 3,38 événements/patient/an. L'incidence des événements d'hypoglycémie sévère a été de 0,7 % et le taux a été de 0,01 événement/patient/an.
Les incidences ont été de 85,3 % et de 80,0 % avec le dulaglutide aux doses respectives de 0,75 mg et de 1,5 mg, utilisé en association avec de l'insuline prandiale, et les taux ont été de 35,66 et 31,06 événements/patient/an. L'incidence des événements d'hypoglycémie sévère a été de 2,4 et de 3,4 % et les taux de 0,05 et de 0,06 événement/patient/an.
Dans une étude de phase 3 d'une durée de 52 semaines, lorsque le dulaglutide aux doses de 1,5 mg, 3 mg et 4,5 mg était utilisé en association à la metformine, les incidences d'hypoglycémie symptomatique documentée ont été respectivement de 3,1 %, 2,4 % et 3,1 %, et les taux ont été de 0,07, 0,05 et 0,07 événements/patient/an. Un épisode d'hypoglycémie sévère a été rapporté avec le dulaglutide aux doses de 1,5 mg et 4,5 mg.
Réactions indésirables gastro-intestinales
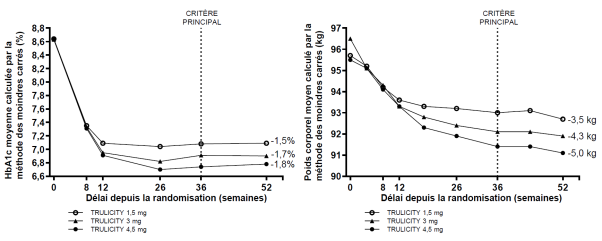
Les évènements indésirables gastro-intestinaux rapportés cumulés sur une durée de 104 semaines avec une dose de dulaglutide de 0,75 mg et 1,5 mg incluaient respectivement : nausées (12,9 % et 21,2 %), diarrhées (10,7 % et 13,7 %) et vomissements (6,9 % et 11,5 %). Ces évènements ont été généralement d'intensité légère à modérée et principalement observés au cours des 2 premières semaines de traitement ; ils ont rapidement diminué au cours des 4 semaines suivantes, période après laquelle le taux est resté relativement stable.
Dans une étude de phase 3 avec le dulaglutide aux doses de 1,5 mg, 3 mg et 4,5 mg, les évènements indésirables gastro-intestinaux rapportés cumulés sur une durée de 52 semaines, incluaient respectivement : nausées (14,2 %, 16,1 % et 17,3 %), diarrhées (7,7 %, 12,0 % et 11,6 %) et vomissements (6,4 %, 9,1 % et 10,1 %).
Lors des études de pharmacologie clinique réalisées chez des patients ayant un diabète de type 2 pendant une durée maximale de 6 semaines, la majorité des évènements indésirables gastro-intestinaux ont été observés au cours des 2 à 3 premiers jours après la dose initiale et ont diminué avec les doses suivantes.
Pancréatite aiguë
L'incidence des pancréatites aiguës dans les études d’enregistrement de phases 2 et 3 a été de 0,07 % pour le dulaglutide versus 0,14 % pour le placebo et 0,19 % pour les comparateurs avec ou sans autre traitement antidiabétique concomitant de fond. Des pancréatites aiguës et des pancréatites ont également été rapportées après commercialisation.
Enzymes pancréatiques
Le dulaglutide est associé à des augmentations moyennes des enzymes pancréatiques par rapport à la valeur à l’inclusion (lipase et/ou amylase pancréatique) de 11 à 21 % (voir rubrique 4.4). En l'absence d'autres signes et symptômes de pancréatite aiguë, des élévations des enzymes pancréatiques seules ne sont pas prédictives d'une pancréatite aiguë.
Augmentation de la fréquence cardiaque
De légères augmentations de la fréquence cardiaque de 2 à 4 battements par minute (bpm) en moyenne et une incidence de 1,3 % et de 1,4 % de tachycardie sinusale, avec une augmentation concomitante ≥ 15 bpm par rapport à la valeur à l’inclusion, ont été observées avec le dulaglutide respectivement à la dose de 0,75 mg et de 1,5 mg.
Dans une étude de phase 3 avec le dulaglutide aux doses de 1,5 mg, 3 mg et 4,5 mg, l’incidence de tachycardie sinusale, avec une augmentation concomitante ≥ 15 bpm par rapport à la valeur à l’inclusion, était respectivement de 2,6 %, 1,9 % et 2,6 %. Des augmentations moyennes de la fréquence cardiaque de 1 à 4 bpm ont été observées.
Bloc auriculo-ventriculaire de premier degré/allongement de l'intervalle PR
De légères augmentations de l'intervalle PR de 2 à 3 msec en moyenne par rapport à la valeur à l’inclusion et une incidence de 1,5 % et de 2,4 % des blocs auriculo-ventriculaire de premier degré ont été observées avec le dulaglutide respectivement à la dose de 0,75 mg et de 1,5 mg.
Dans une étude de phase 3 avec le dulaglutide aux doses de 1,5 mg, 3 mg et 4,5 mg, l’incidence des blocs auriculo-ventriculaire de premier degré a été respectivement de 1,2 %, 3,8 % et 1,7 %. Des augmentations de l'intervalle PR de 3 à 5 msec en moyenne par rapport à la valeur à l’inclusion ont été observées.
Immunogénicité
Lors des études d’enregistrement, le traitement par dulaglutide a été associé à une incidence de 1,6 % d’apparition d'anticorps anti‑médicament dulaglutide, ce qui indique que les modifications structurelles des portions GLP‑1 et IgG4 modifiées de la molécule dulaglutide, ainsi que la forte homologie avec le GLP‑1 et l'IgG4 natifs, minimisent le risque de réponse immunitaire contre le dulaglutide. Les patients développant des anticorps anti‑médicament dulaglutide présentaient généralement des titres faibles ; et bien que le nombre de patients développant des anticorps anti‑médicament dulaglutide ait été limité, l'examen des données de phase 3 ne montre pas d’impact avéré des anticorps anti‑médicament dulaglutide sur les changements de l'HbA1c. Aucun des patients ayant présenté une hypersensibilité systémique n'a développé d'anticorps anti‑médicament dulaglutide.
Hypersensibilité
Lors des études d’enregistrement de phases 2 et 3, des événements d'hypersensibilité systémique (par ex., urticaire, œdème) ont été signalés chez 0,5 % des patients traités par dulaglutide. De rares cas de réactions anaphylactiques ont été rapportés avec l’utilisation du dulaglutide commercialisé.
Réactions au site d'injection
Des réactions au site d'injection ont été signalées chez 1,9 % des patients traités par dulaglutide. Des réactions indésirables au site d'injection à médiation immunitaire potentielle (par ex., éruption cutanée, érythème) ont été signalées chez 0,7 % des patients ; elles ont été généralement d'intensité légère.
Arrêt du traitement suite à un évènement indésirable
Lors des études de 26 semaines, la fréquence des arrêts de traitement suite à des évènements indésirables a été de 2,6 % (0,75 mg) et de 6,1 % (1,5 mg) pour le dulaglutide versus 3,7 % pour le placebo. Pendant toute la durée de l'étude (104 semaines maximum), la fréquence des arrêts de traitement suite à des évènements indésirables a été de 5,1 % (0,75 mg) et de 8,4 % (1,5 mg) pour le dulaglutide. Les réactions indésirables entraînant le plus fréquemment un arrêt du traitement pour le dulaglutide 0,75 mg et 1,5 mg étaient respectivement les nausées (1,0 % ; 1,9 %), les diarrhées (0,5 % ; 0,6 %) et les vomissements (0,4 % ; 0,6 %) ; et ont été généralement rapportées au cours des 4 à 6 premières semaines.
Dans une étude de phase 3 avec le dulaglutide aux doses de 1,5 mg, 3 mg et 4,5 mg, la fréquence des arrêts de traitement suite à des évènements indésirables a été de 6,0 % (1,5 mg), 7,0 % (3 mg) et 8,5 % (4,5 mg) sur une durée de 52 semaines. Les réactions indésirables entraînant le plus fréquemment un arrêt du traitement pour le dulaglutide 1,5 mg, 3 mg et 4,5 mg étaient respectivement les nausées (1,3 %, 1,3 % ; 1,5 %), les diarrhées (0,2 % ; 1,0 % ; 1,0 %) et les vomissements (0,0 % ; 0,8 % ; 1,3 %).
Dulaglutide aux doses de 3 mg et 4,5 mg
Le profil de sécurité observé chez les patients traités par dulaglutide aux doses hebdomadaires de 3 mg et 4,5 mg est cohérent avec celui décrit ci-dessus pour le dulaglutide aux doses hebdomadaires de 0,75 mg et 1,5 mg.
Population pédiatrique
Le profil de sécurité observé chez les patients pédiatriques âgés de 10 ans et plus traités par dulaglutide aux doses hebdomadaires de 0,75 mg et 1,5 mg est comparable avec celui décrit ci-dessus pour les patients adultes.
Le profil d'immunogénicité chez les patients pédiatriques traités par dulaglutide est cohérent avec celui décrit ci-dessus pour les patients adultes. Dans l'étude pédiatrique, 2,1 % et 4,0 % des patients traités respectivement par placebo et par dulaglutide ont développé des anticorps anti-médicament dulaglutide.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via :
Belgique :
Agence fédérale des médicaments et des produits de santé, www.afmps.be, Division Vigilance: Site internet: www.notifieruneffetindesirable.be, e-mail: adr@fagg-afmps.be.
Luxembourg :
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet : www.guichet.lu/pharmacovigilance.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Eli Lilly Nederland BV., Orteliuslaan 1000, 3528 BD Utrecht, Pays-Bas.
8. NUMÉROS D'AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/14/956/001
EU/1/14/956/002
EU/1/14/956/003
EU/1/14/956/006
EU/1/14/956/007
EU/1/14/956/008
EU/1/14/956/011
EU/1/14/956/012
EU/1/14/956/013
EU/1/14/956/014
EU/1/14/956/015
EU/1/14/956/016
10. DATE DE MISE A JOUR DU TEXTE : 30 JANVIER 2026
STATUT LEGAL DE DELIVRANCE Médicament sur prescription médicale.
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3275971 | TRULICITY 0,75MG/0,5ML SOL INJ STYLO PREREMPLI 4 | A10BJ05 | € 104,05 | - | Oui | € 2 | € 1 |
| 3275989 | TRULICITY 1,50MG/0,5ML SOL INJ STYLO PREREMPLI 4 | A10BJ05 | € 104,05 | - | Oui | € 2 | € 1 |