1. DÉNOMINATION DU MÉDICAMENT
Invokana 100 mg comprimés pelliculés
Invokana 300 mg comprimés pelliculés
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Invokana 100 mg comprimés pelliculés
Chaque comprimé contient de l’hémihydrate de canagliflozine, équivalent à 100 mg de canagliflozine.
Excipient(s) à effet notoire:
Chaque comprimé contient 39,26 mg de lactose.
Invokana 300 mg comprimés pelliculés
Chaque comprimé contient de l’hémihydrate de canagliflozine, équivalent à 300 mg de canagliflozine.
Excipient(s) à effet notoire:
Chaque comprimé contient 117,78 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé).
Invokana 100 mg comprimés pelliculés
Comprimé jaune, en forme de gélule, d’environ 11 mm de longueur, à libération immédiate et pelliculé, avec l’inscription « CFZ » sur une face et « 100 » sur l’autre face.
Invokana 300 mg comprimés pelliculés
Comprimé blanc, en forme de gélule, d’environ 17 mm de longueur, à libération immédiate et pelliculé, avec l’inscription « CFZ » sur une face et « 300 » sur l’autre face.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Invokana est indiqué dans le traitement des adultes et des enfants agés de 10 ans et plus atteints de diabète de type 2 insuffisamment contrôlé, en complément du régime alimentaire et de l’exercice physique :
- En monothérapie quand la metformine est considérée comme étant inappropriée en raison d’une intolérance ou de contre-indications
- En complément d’autres médicaments pour le traitement du diabète.
Concernant les résultats d’études vis-à-vis des associations de traitements, des effets sur le contrôle glycémique des événements cardiovasculaires et rénaux et des populations étudiées, voir les rubriques 4.4, 4.5 et 5.1.
4.2 Posologie et mode d’administration
Posologie
La dose initiale de canagliflozine recommandée est de 100 mg une fois par jour. Chez les patients qui tolèrent la dose de 100 mg de canagliflozine une fois par jour, et dont le débit de filtration glomérulaire estimé (DFGe) est ≥ 60 mL/min/1,73 m² ou la ClCr ≥ 60 mL/min et qui nécessitent un contrôle glycémique plus étroit, la dose peut être augmentée à 300 mg par jour (voir rubrique 4.4). Pour les recommandations concernant l’adaptation posologique en fonction du DFGe, consulter le tableau 1.
Des précautions doivent être prises lors de l’augmentation de la dose chez les patients âgés de 75 ans et plus, les patients atteints d’une maladie cardiovasculaire, ou les autres patients pour lesquels la diurèse initiale induite par la canagliflozine présente un risque (voir rubrique 4.4). Chez les patients présentant des signes de déplétion volémique, il est recommandé de corriger cet état avant l’instauration du traitement par la canagliflozine (voir rubrique 4.4).
Lorsque la canagliflozine est utilisée en association à l’insuline ou à un sécrétagogue de l’insuline (par exemple les sulfamides hypoglycémiants), une dose plus faible d’insuline ou de sécrétagogue de l’insuline peut être envisagée pour réduire le risque d’hypoglycémie (voir rubriques 4.5 et 4.8).
Populations particulières
Patients âgés
La fonction rénale et le risque de déplétion volémique doivent être pris en compte (voir rubrique 4.4).
Insuffisance rénale
Pour le traitement de la maladie rénale chronique chez les patients diabétiques de type 2, en complément du traitement standard (par exemple des IEC ou des ARA II), une dose de 100 mg de canagliflozine une fois par jour doit être utilisée (voir tableau 1). Du fait que l’efficacité hypoglycémiante de la canagliflozine est réduite chez les patients présentant une insuffisance rénale modérée et probablement absente chez les patients présentant une insuffisance rénale sévère, si un contrôle glycémique renforcé est nécessaire, l’ajout d’agents anti-hyperglycémiques doit être envisagé. Pour les recommandations concernant l’adaptation posologique en fonction du DFGe, consulter le tableau 1.
Tableau 1 : Recommandations concernant l’adaptation posologique chez les adultes et les enfants âgés de 10 ans et plusa | |
DFGe (mL/min/1,73 m2) | Dose quotidienne totale de canagliflozine |
≥ 60 | Initier avec 100 mg. |
30 à < 60b | Utiliser 100 mg. |
< 30b,c | Continuer avec 100 mg pour les patients prenant déjà Invokanad. |
a Voir rubriques 4.4, 4.8, 5.1 et 5.2. | |
Insuffisance hépatique
Chez les patients atteints d’insuffisance hépatique légère ou modérée, aucune adaptation posologique n’est nécessaire.
La canagliflozine n’a pas été étudiée chez les patients présentant une insuffisance hépatique sévère et son utilisation n’est pas recommandée chez ces patients (voir rubrique 5.2).
Population pédiatrique
Aucun ajustement posologique n’est nécessaire pour le diabète de type 2 chez les enfants âgés de 10 ans et plus (voir rubriques 5.1 et 5.2). Chez les enfants pesant < 50 kg, la prudence est recommandée lors de l'augmentation de la dose jusqu'à 300 mg, car les données de sécurité sont limitées (voir rubrique 4.4).
La sécurité et l’efficacité d’Invokana n’ont pas été établies chez les enfants de moins de 10 ans.
Mode d’administration
Voie orale.
Invokana doit être pris par voie orale une fois par jour, de préférence avant le premier repas de la journée. Les comprimés doivent être avalés entiers.
Si une dose est oubliée, elle doit être prise dès que le patient s’en souvient ; cependant, aucune dose double ne doit être prise le même jour.
4.3 Contre-indications
- Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
La sécurité de la canagliflozine a été évaluée chez 22 645 patients adultes atteints de diabète de type 2, dont 13 278 patients traités par canagliflozine et 9 367 par un comparateur lors de 15 études cliniques contrôlées de phase 3 et 4 menées en double-aveugle. Au total, 10 134 patients adultes ont été traités dans le cadre de deux études cardiovasculaires dédiées, pendant une durée moyenne d’exposition de 149 semaines (223 semaines dans l’essai CANVAS et 94 semaines dans l’essai CANVAS-R) et 8 114 patients adultes ont été traités lors de 12 études cliniques contrôlées de phase 3 et 4 menées en double-aveugle pendant une durée moyenne d’exposition de 49 semaines. Dans une étude dédiée du devenir de la fonction rénale, un total de 4 397 patients adultes atteints de diabète de type 2 et de maladie rénale chronique ont eu une durée moyenne d’exposition de 115 semaines.
L’évaluation principale de la sécurité et de la tolérance a été effectuée dans une analyse poolée (n = 2 313) de quatre études cliniques contrôlées versus placebo de 26 semaines (en monothérapie et en association à metformine, à metformine + sulfamide hypoglycémiant et à metformine + pioglitazone) chez les adultes. Les effets indésirables les plus fréquemment rapportés pendant le traitement ont été l’hypoglycémie, lors de l’association à l’insuline ou à un sulfamide hypoglycémiant, les candidoses vulvovaginales, les infections des voies urinaires, ainsi que la polyurie ou la pollakiurie (mictions plus abondantes et plus fréquentes). Les effets indésirables conduisant à l’arrêt du traitement chez ≥ 0,5 % de l’ensemble des patients adultes traités par la canagliflozine dans ces études ont été des candidoses vulvovaginales (0,7 % des femmes traitées), ainsi que des balanites ou des balanoposthites (0,5 % des hommes traités). D’autres analyses de sécurité (incluant des données à long terme) ont été effectuées sur les données correspondant à l’ensemble du programme d’études de la canagliflozine (études contrôlées versus placebo et versus comparateur actif) pour évaluer les effets indésirables rapportés, afin d’identifier les effets indésirables (tableau 2) (voir rubriques 4.2 et 4.4).
Tableau des effets indésirables
Les effets indésirables présentés dans le tableau 2 sont issus des analyses poolées des études menées versus placebo et comparateur actif décrites ci-dessus. Les effets indésirables rapportés par l’utilisation post-commercialisation de la canagliflozine dans le monde entier sont aussi inclus dans ce tableau. Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classe de systèmes d’organes. Les différentes catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 2 : Tableau des effets indésirables (MedDRA) à partir des études contrôlées versus placeboe et comparateur actif ainsi que de l’expérience post-commercialisation | ||
Classe de systèmes d’organes | Effet indésirable | |
Infections et infestations | ||
Très fréquent | Candidose vulvovaginaleb,j | |
Fréquent | Balanite ou balanoposthiteb,k, Infection des voies urinairesc (pyélonéphrite et sepsis urinaire ont été rapportés après la commercialisation) | |
Indéterminée | Fasciite nécrosante du périnée (gangrène de Fournier)d | |
Affections du système immunitaire | ||
Rare | Réaction anaphylactique | |
Troubles du métabolisme et de la nutrition | ||
Très fréquent | Hypoglycémie en association à l’insuline ou à un sulfamide hypoglycémiantc | |
Peu fréquent | Déshydratationa | |
Rare | Acidocétose diabétique,b | |
Affections du système nerveux | ||
Peu fréquent | Sensation vertigineuse posturalea, Syncopea | |
Affections vasculaires | ||
Peu fréquent | Hypotensiona, Hypotension orthostatiquea | |
Affections gastro-instestinales | ||
Fréquent | Constipation, Soiff, Nausées | |
Affections de la peau et du tissu sous-cutané | ||
Peu fréquent | Photosensibilité, Éruption cutanéeg, Urticaire | |
Rare | Angiœdème | |
Affections musculo-squelettiques et systémiques | ||
Peu fréquent | Fracture osseuseh | |
Affection du rein et des voies urinaires | ||
Fréquent | Polyurie ou Pollakiuriei | |
Peu fréquent | Insuffisance rénale (principalement dans le contexte de déplétion volémique) | |
Investigations | ||
Fréquent | Dyslipidémiel, Hématocrite augmenté,b,m | |
Peu fréquent | Créatininémie augmentée,b,n, Urémie augmentée,b,o, kaliémie augmentée,b,p, Phosphatémie augmentéeq | |
Actes médicaux et chirurgicaux | ||
Peu fréquent | Amputation des membres inférieurs (principalement l’orteil et du médio pied), en particulier chez les patients à haut risque de maladie cardiaqueb | |
a Liées à une déplétion volémique ; voir rubrique 4.4 et la description de l’effet indésirable (EI) ci-dessous. | ||
Description de certains effets indésirables
Acidocétose diabétique
Dans une étude du devenir de la fonction rénale à long terme chez des patients adultes atteints de diabète de type 2 et de maladie rénale chronique, les taux d’incidence des événements avérés d’acidocétose diabétique (ACD) étaient de 0,21 (0,5 %, 12/2 200) et de 0,03 (0,1 %, 2/2 197) pour 100 patients-années de suivi avec la canagliflozine 100 mg et le placebo, respectivement ; parmi les 14 patients atteints d’ACD, 8 (7 sous canagliflozine 100 mg et 1 sous placebo) avaient un DFGe avant traitement, compris entre 30 et ˂ 45 mL/min/1,73 m2 (voir rubrique 4.4).
Amputation des membres inférieurs
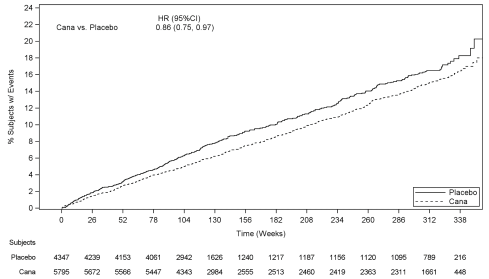
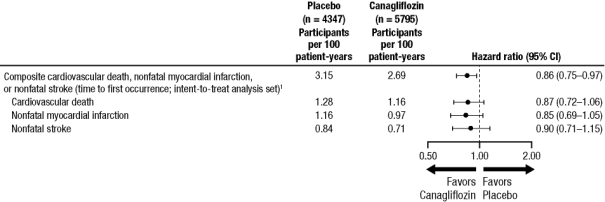
Chez les patients souffrant de diabète de type 2 et présentant une maladie cardiovasculaire établie ou au moins 2 facteurs de risque de maladie cardiovasculaire, la canagliflozine a été associée à une augmentation du risque d’amputation des membres inférieurs, tel qu’observé dans le programme intégré CANVAS, comprenant CANVAS et CANVAS-R, deux essais de grande ampleur, de longue durée, randomisés, contrôlés contre placebo visant à évaluer 10 134 patients adultes. Le déséquilibre s’est produit dés les 26 premières semaines de traitement. Les patients des essais CANVAS et CANVAS-R ont été suivis pendant une moyenne de respectivement 5,7 et 2,1 ans. Indépendamment du fait que le traitement soit la canagliflozine ou le placebo, le risque d’amputation était supérieur chez les patients ayant un antécédent d’amputation, de maladie vasculaire périphérique ou de neuropathie. Le risque d’amputation des membres inférieurs n’était pas dose-dépendant. Les résultats relatifs à l’amputation du programme intégré CANVAS sont présentés par le tableau 3.
Il n’y avait pas de différence de risque d’amputation des membres inférieurs associé à l’utilisation de canagliflozine 100 mg par rapport au placebo (1,2 contre 1,1 événements pour 100 patients-années, respectivement [RR : 1,11 ; IC 95 % 0,79, 1,56]) dans l’étude CREDENCE, une étude du devenir de la fonction rénale à long terme de 4 397 patients adultes atteints de diabète de type 2 et de maladie rénale chronique (voir rubrique 4.4). Dans d’autres études sur le diabète de type 2 portant sur la canagliflozine, incluant une population diabétique générale de 8 114 patients adultes, aucune différence n’a été observée par rapport au contrôle vis-à-vis du risque d’amputation des membres inférieurs.
Tableau 3 : Analyse intégrée des amputations dans les essais CANVAS ET CANVAS-R | ||
| Placebo | canagliflozine |
Nombre total de sujets présentant des événements, n (%) | 47 (1,1) | 140 (2,4) |
Taux d’incidence (pour 100 patients-années) | 0,34 | 0,63 |
RR (IC 95 %) vs. placebo |
| 1,97 (1,41, 2,75) |
Amputation mineure, n (%)* | 34/47 (72,3) | 99/140 (70,7) |
Amputation majeure, n (%)† | 13/47 (27,7) | 41/140 (29,3) |
Remarque : l’incidence repose sur le nombre de patients ayant subi au moins une amputation et non le nombre total d’événements d’amputations. Le suivi d’un patient est calculé à partir du Jour 1 jusqu’à la date de la première amputation. Certains patients ont subi plus d’une amputation. Le pourcentage d’amputations mineures et majeures se fonde sur le niveau d’amputation le plus élevé pour chaque patient. | ||
Dans le programme CANVAS, chez les sujets ayant subi une amputation, l’orteil et le médio pied représentaient les sites les plus fréquemment touchés (71%) dans les deux groupes de traitement (tableau 3). Des amputations multiples (certaines impliquant les deux membres inférieurs) ont été observées peu fréquemment et dans des proportions similaires au sein des deux groupes de traitement.
Les infections des membres inférieurs, ulcères du pied diabétique, artériopathies périphériques et gangrènes étaient les événements médicaux les plus fréquemment associés à la nécessité d’une amputation au sein des deux groupes de traitement (voir rubrique 4.4).
Effets indésirables liés à la déplétion volémique
Dans les analyses poolées des quatre études contrôlées versus placebo de 26 semaines chez les adultes, l’incidence de tous les effets indésirables liés à la déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension, déshydratation et syncope) a été de 1,2 % pour canagliflozine 100 mg, 1,3 % pour canagliflozine 300 mg et 1,1 % pour le placebo. Dans les deux études contrôlées versus traitement actif, l’incidence avec la canagliflozine a été similaire à celle observée avec les comparateurs actifs.
Dans l’une des études cardiovasculaires de longue durée dédiées (CANVAS), dans laquelle les patients adultes étaient généralement plus âgés, avec un taux plus élevé de complications diabétiques, les taux d’incidence des effets indésirables liés à la déplétion volémique ont été de 2,3 événements pour 100 patients-années avec canagliflozine 100 mg une fois par jour, 2,9 avec canagliflozine 300 mg et 1,9 avec placebo.
Pour évaluer les facteurs de risque relatifs à ces effets indésirables, une analyse poolée à plus grande échelle (N = 12 441) a été menée chez des patients adultes provenant de 13 études de phase 3 et de phase 4 contrôlées incluant les deux doses de canagliflozine. Dans cette analyse poolée, les patients traités par diurétiques de l’anse, les patients avec un DFGe initial compris entre 30 ml/min/1,73 m2 et < 60 ml/min/1,73 m2 et les patients âgés de 75 ans et plus avaient généralement des incidences supérieures de ces effets indésirables. Pour les patients sous diurétiques de l’anse, les taux d’incidence ont été de 5,0 événements pour 100 patients-années avec canagliflozine 100 mg et 5,7 avec canagliflozine 300 mg, contre 4,1 événements pour 100 patients-années d’exposition dans le groupe contrôle. Pour les patients avec un DFGe initial compris entre 30 ml/min/1,73 m2 et < 60 ml/min/1,73 m2, les taux d’incidence ont été de 5,2 événements pour 100 patients-années d’exposition avec canagliflozine 100 mg et 5,4 avec canagliflozine 300 mg, contre 3,1 événements pour 100 patients-années d’exposition dans le groupe témoin. Chez les patients âgés de 75 ans et plus, les taux d’incidence ont été de 5,3 avec canagliflozine 100 mg et 6,1 avec canagliflozine 300 mg, contre 2,4 événements pour 100 patients-années d’exposition dans le groupe contrôle (voir rubriques 4.2 et 4.4).
Dans une étude du devenir de la fonction rénale à long terme chez des patients adultes atteints de diabète de type 2 et de maladie rénale chronique, les taux d’incidence des événements liés à la déplétion volémique étaient de 2,84 et 2,35 événements pour 100 patients-années pour canagliflozine 100 mg et le placebo, respectivement. Il a été observé que le taux d’incidence augmentait lorsque le DFGe diminuait. Chez les sujets présentant un DFGe compris entre 30 et <45 mL/min/1,73 m2, le taux d’incidence de la déplétion volémique était plus élevé dans le groupe canagliflozine (4,91 événements pour 100 patients-années) que dans le groupe placebo (2,60 événements pour 100 patients-années) ; toutefois, dans les sous-groupes ayant un DFGe compris entre ≥45 et <60 et compris entre 60 et <90 mL/min/1,73 m2, le taux d’incidence entre les groupes était similaire.
Dans l’étude cardiovasculaire dédiée et l’analyse poolée à plus grande échelle, ainsi que dans l’étude chez l’adulte dédiée du devenir de la fonction rénale, les arrêts de traitements dus à des effets indésirables liés à la déplétion volémique et à des effets indésirables graves liés à la déplétion volémique n’ont pas augmenté avec la canagliflozine.
Hypoglycémie en cas d’association à l’insuline ou à un sécrétagogue de l’insuline
La fréquence des hypoglycémies a été faible (environ 4 %) dans les différents groupes de traitement, y compris le groupe sous placebo, lorsque la canagliflozine a été utilisée en monothérapie ou en association à la metformine. Lorsque la canagliflozine a été ajoutée à une insulinothérapie, on a observé une hypoglycémie chez respectivement 49,3 %, 48,2 % et 36,8 % des patients adultes traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo et une hypoglycémie sévère s’est produite chez respectivement 1,8 %, 2,7 % et 2,5 % des patients adultes traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo. Lorsque la canagliflozine a été ajoutée à un traitement par sulfamide hypoglycémiant, une hypoglycémie a été observée chez respectivement 4,1 %, 12,5 % et 5,8 % des patients adultes traités par canagliflozine 100 mg, canagliflozine 300 mg et placebo (voir rubriques 4.2 et 4.5).
Infections mycosiques génitales
Une candidose vulvovaginale (incluant une vulvovaginite et une infection mycosique vulvovaginale) a été observée chez respectivement 10,4 % et 11,4 % des femmes adultes traitées par canagliflozine 100 mg et canagliflozine 300 mg, contre 3,2 % chez les patientes sous placebo. La plupart des candidoses vulvovaginales sont apparues au cours des quatre premiers mois de traitement par canagliflozine. 2,3 % des femmes sous canagliflozine ont présenté plus d’une infection. Dans l’ensemble, 0,7 % des patientes ont arrêté le traitement par canagliflozine en raison d’une candidose vulvovaginale. La durée des symptômes était comparable entre le traitement par canagliflozine et le placebo (voir rubrique 4.4). Dans le programme CANVAS, les patients souffrant de diabète de type 2 et présentant une maladie cardiovasculaire établie ou au moins 2 facteurs de risques de maladie cardiovasculaire ont subi une durée d’infection plus longue (voir rubrique 4.4). Dans le programme CANVAS, la durée médiane de l’infection était plus longue dans le groupe canagliflozine que dans le groupe placebo.
Une balanite candidosique ou une balanoposthite a affecté les hommes adultes selon un taux de, respectivement, 2,98 et 0,79 événements pour 100 patients-années sous canagliflozine et placebo. Chez les hommes sous canagliflozine, 2,4 % ont présenté plus d’une infection. L’arrêt de la canagliflozine chez les hommes en raison d’une balanite candidosique ou d’une balanoposthite s’est produit selon un taux de 0,37 événements pour 100 patients-années. Des phimosis ont été rapportés selon un taux de, respectivement, 0,39 et 0,07 événements pour 100 patients-années sous canagliflozine et placebo. Une circoncision a été réalisée respectivement à des taux de 0,31 et 0,09 événements pour 100 patients-années sous canagliflozine et placebo (voir rubrique 4.4).
Infections des voies urinaires
Dans les études cliniques chez les adultes, la survenue d’infections des voies urinaires a été plus fréquente sous canagliflozine 100 mg et 300 mg (respectivement 5,9 %, et 4,3 %), comparativement à la fréquence observée sous placebo (4,0 %). La plupart des infections ont été légères à modérées, sans augmentation de l’apparition des effets indésirables graves. Dans ces études, les sujets ont répondu à des traitements standards tout en continuant le traitement par canagliflozine.
Néanmoins, des cas d’infection des voies urinaires compliquée, y compris de pyélonéphrite et de sepsis urinaire, survenus après mise sur le marché ont été rapportés chez des patients traités par canagliflozine et ont fréquemment mené à l’interruption du traitement.
Fracture osseuse
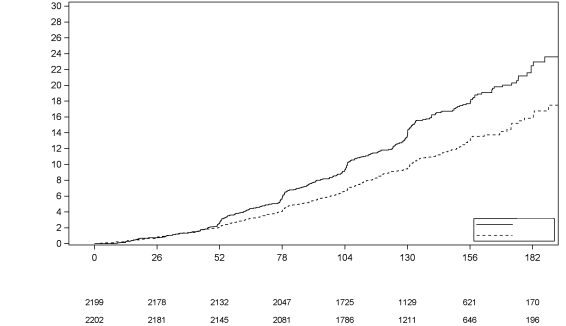
Dans une étude cardiovasculaire (CANVAS) de 4 327 sujets adultes traités pour une maladie cardiovasculaire établie ou présentant au moins deux facteurs de risque de maladie cardiovasculaire, les taux d’incidence de l’ensemble des fractures osseuses avérées étaient respectivement de 1,6, 1,8 et 1,1 événements pour 100 patients-années de suivi, respectivement sous 100 mg de canagliflozine, 300 mg de canagliflozine et placebo, avec un déséquilibre de ce taux survenant initialement dans les 26 premières semaines de traitement.
Dans deux autres études à long terme chez les adultes et dans des études chez les adultes effectuées dans la population diabétique générale, aucune différence concernant le risque de fracture n’a été observée avec la canagliflozine par rapport au groupe contrôle. Dans une deuxième étude cardiovasculaire (CANVAS-R) de 5 807 sujets adultes traités pour une maladie cardiovasculaire établie ou présentant au moins deux facteurs de risque de maladie cardiovasculaire, les taux d’incidence de l’ensemble des fractures osseuses avérées étaient respectivement de 1,1 et 1,3 événements pour 100 patients-années de suivi, sous canagliflozine et placebo.
Dans une étude du devenir de la fonction rénale à long terme de 4 397 sujets adultes traités atteints de diabète de type 2 et de maladie rénale chronique, les taux d’incidence de l’ensemble des fractures osseuses avérées étaient de 1,2 événements pour 100 patients-années de suivi avec la canagliflozine 100 mg et avec le placebo. Dans les autres études avec la canagliflozine dans le diabète de type 2, qui ont inclus une population diabétique générale de 7 729 patients adultes et où les fractures osseuses ont été évaluées, les taux d’incidence de l’ensemble des fractures osseuses avérées étaient respectivement de 1,2 et 1,1 événements pour 100 patients-années de suivi, sous canagliflozine et placebo. Après 104 semaines de traitement, la canagliflozine n’a pas affecté la densité minérale osseuse.
Populations particulières
Patients âgés
Dans une analyse poolée de 13 études contrôlées versus placebo et contrôlées versus comparateur actif, le profil de sécurité de canagliflozine chez les patients âgés était généralement cohérent avec celui des patients plus jeunes. Les patients âgés de 75 ans et plus avaient une incidence plus élevée d’effets indésirables liés à la déplétion volémique (comme les sensations vertigineuses posturales, l’hypotension orthostatique, l’hypotension), avec des taux d’incidence respectivement de 5,3, 6,1 et 2,4 événements pour 100 patients-années d’exposition pour canagliflozine 100 mg, canagliflozine 300 mg et le groupe contrôle. Des diminutions du DFGe (-3,4 et -4,7 ml/min/1,73 m2) ont été respectivement rapportées dans les groupes canagliflozine 100 mg et 300 mg, comparativement au groupe contrôle (‑4.2 ml/min/1,73 m2). Les DFGe moyens de référence étaient respectivement de 62,5, 64,7 et 63,5 ml/min/1,73 m2 pour la canagliflozine 100 mg, la canagliflozine 300 mg et le groupe contrôle (voir rubriques 4.2 et 4.4).
Insuffisance rénale chez les patients adultes atteints de diabète de type 2 insuffisamment contrôlé
Les patients adultes avec un DFGe < 60 ml/min/1,73 m2 avaient une incidence supérieure d’effets indésirables associés à la déplétion volémique (par exemple, sensation vertigineuse posturale, hypotension orthostatique, hypotension) avec des taux d’incidence respectivement de 5,3, 5,1 et 3,1 événements pour 100 patients-années d’exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo (voir rubriques 4.2 et 4.4).
Le taux d’incidence globale de potassium sérique élevé était supérieur chez les patients souffrant d’insuffisance rénale modérée, avec des taux d’incidence, respectivement, de 4,9, 6,1 et 5,4 événements pour 100 patients-années d’exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo. En général, ces augmentations de la kaliémie ont été transitoires et n’ont pas nécessité de traitement spécifique.
Chez les patients souffrant d’insuffisance rénale modérée, une augmentation de la créatinine sérique de 9,2 µmol/l et de l’azote uréique du sang d’environ 1,0 mmol/l aux deux doses de canagliflozine a été observée.
Les taux d’incidence de plus fortes diminutions du DFGe (> 30 %), à tout moment du traitement, étaient respectivement de 7,3, 8,1 et 6,5 événements pour 100 patients-années d’exposition pour canagliflozine 100 mg, canagliflozine 300 mg et placebo. À la dernière valeur post-référence, les taux d’incidence de ces diminutions étaient de 3,3 pour les patients traités par canagliflozine 100 mg, 2,7 pour canagliflozine 300 mg et 3,7 événements pour 100 patients-années d’exposition pour le placebo (voir rubrique 4.4).
Quel que soit leur DFGe de référence, chez les patients traités par canagliflozine, le DFGe moyen a commencé par chuter. Par la suite, le DFGe s’est maintenu ou a progressivement augmenté durant la suite du traitement. Le DFGe moyen est revenu à la valeur de référence après l’arrêt du traitement, ce qui suggère que des modifications hémodynamiques peuvent jouer un rôle dans ces modifications de la fonction rénale.
Insuffisance rénale chez les patients adultes atteints de maladie rénale chronique dans le diabète de type 2
Dans une étude du devenir de la fonction rénale à long terme chez des patients adultes atteints de diabète de type 2 et de maladie rénale chronique, l’incidence des événements liés à la fonction rénale était élevée dans les deux groupes, mais moins élevée dans le groupe canagliflozine (5,71 événements pour 100 patients-années) que dans le groupe placebo (7,91 événements pour 100 patients-années). Les événements graves et sévères liés à la fonction rénale étaient également moins fréquents dans le groupe canagliflozine que dans le groupe placebo. Les taux d’incidence des événements liés à la fonction rénale étaient inférieurs avec la canagliflozine par rapport au placebo dans les trois strates de DFGe ; le taux d’incidence le plus élevé des événements liés à la fonction rénale a été observé dans la strate où le DFGe est compris entre 30 et < 45 mL/min/1,73 m2 (9,47 contre 12,80 événements pour 100 patients-années respectivement pour la canagliflozine et le placebo).
Dans l’étude du devenir de la fonction rénale à long terme, aucune différence du potassium sérique, aucune augmentation des événements indésirables d’hyperkaliémie, et aucune augmentation absolue (> 6,5 mEq/L) ou relative (> limite supérieure de la normale et > 15 % d’augmentation par rapport aux valeurs initiales) du potassium sérique n’ont été observées avec canagliflozine 100 mg par rapport au placebo.
En général, aucun déséquilibre n’a été observé entre les groupes de traitement en ce qui concerne les anomalies du phosphate, dans l’ensemble ou dans chaque catégorie de DFGe (45 à < 60 ou 30 à < 45 mL/min/1,73 m2 [ClCr 45 à < 60 ou 30 à < 45 mL/min]).
Population pédiatrique
Dans l'étude DIA3018, 171 enfants âgés de 10 ans et plus atteints de diabète de type 2 ont reçu un traitement : 84 participants ont reçu de la canagliflozine et 87 ont reçu un placebo (voir rubrique 5.1). Globalement, la fréquence, le type et la sévérité des effets indésirables chez les enfants âgés de 10 ans et plus étaient comparables à ceux observés dans la population adulte. Les événements indésirables apparus sous traitement plus fréquemment avec la canagliflozine qu’avec le placebo chez les enfants ont été les suivants: maux de tête, rhinopharyngite, infection des voies urinaires et vomissements. Des infections génitales mycosiques ou bactériennes ont été signalées chez un petit nombre de patients recevant de la canagliflozine et aucun avec le placebo.
Aucun de ces événements apparus sous traitement n’ont été sévères ou graves
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de la santé déclarent tout effet indésirable suspecté :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet: www.guichet.lu/pharmacovigilance
Pays-Bas
Nederlands Bijwerkingen Centrum Lareb
Site internet: www.lareb.nl





7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgique
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
Invokana 100 mg comprimés pelliculés
EU/1/13/884/001 (10 comprimés pelliculés)
EU/1/13/884/002 (30 comprimés pelliculés)
EU/1/13/884/003 (90 comprimés pelliculés)
EU/1/13/884/004 (100 comprimés pelliculés)
Invokana 300 mg comprimés pelliculés
EU/1/13/884/005 (10 comprimés pelliculés)
EU/1/13/884/006 (30 comprimés pelliculés)
EU/1/13/884/007 (90 comprimés pelliculés)
EU/1/13/884/008 (100 comprimés pelliculés)
10. DATE DE MISE À JOUR DU TEXTE
08/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3091212 | INVOKANA 100 MG TABL PELL 30 X 100 MG | A10BK02 | € 62,95 | - | Oui | € 2 | € 1 |
| 3091220 | INVOKANA 100 MG TABL PELL 90 X 100 MG | A10BK02 | € 137,25 | - | Oui | € 2 | € 1 |
| 3091238 | INVOKANA 300 MG TABL PELL 90 X 300 MG | A10BK02 | € 200,38 | - | Oui | € 2 | € 1 |
| 3091246 | INVOKANA 300 MG TABL PELL 30 X 300 MG | A10BK02 | € 89,89 | - | Oui | € 2 | € 1 |