1. DÉNOMINATION DU MÉDICAMENT
XGEVA 120 mg solution injectable
XGEVA 120 mg solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 120 mg de denosumab dans 1,7 mL de solution (70 mg/mL).
Chaque seringue préremplie contient 120 mg de denosumab dans 1,0 mL de solution (120 mg/mL).
Le denosumab est un anticorps monoclonal IgG2 humain produit dans une lignée cellulaire de mammifère (cellules d’ovaires de hamster chinois) par la technique de l’ADN recombinant.
Excipient à effet notoire :
Chaque 1,7 mL de solution contient 78 mg de sorbitol (E420).
Chaque 1,0 mL de solution contient 37 mg de sorbitol (E420) et 6,1 mg de L‑phénylalanine.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
XGEVA 120 mg solution injectable
Solution injectable.
XGEVA 120 mg solution injectable en seringue préremplie
Solution injectable.
Solution limpide, incolore à légèrement jaune, pouvant contenir des traces de particules protéiques translucides à blanches.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Prévention des complications osseuses (fractures pathologiques, irradiation osseuse, compression médullaire ou chirurgie osseuse) chez des patients adultes présentant une affection maligne avancée avec atteinte osseuse (voir rubrique 5.1).
Traitement des adultes et des adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes, non résécables ou pour lesquels la résection chirurgicale est susceptible d’entraîner une morbidité sévère.
4.2 Posologie et mode d'administration
XGEVA doit être administré sous la responsabilité d’un professionnel de santé.
Posologie
Une supplémentation quotidienne apportant au moins 500 mg de calcium et 400 UI de vitamine D est requise chez tous les patients, sauf en cas d’hypercalcémie (voir rubrique 4.4).
Les patients traités par XGEVA devront recevoir la notice et la carte d’information au patient.
Prévention des complications osseuses chez les patients adultes présentant une affection maligne avancée avec atteinte osseuse
La posologie recommandée est de 120 mg, administrée une fois toutes les quatre semaines, par injection sous‑cutanée dans la cuisse, l’abdomen ou le bras.
Tumeur osseuse à cellules géantes
La posologie recommandée est de 120 mg de XGEVA toutes les quatre semaines administrée en une seule injection par voie sous‑cutanée dans la cuisse, l’abdomen ou le bras avec une dose supplémentaire de 120 mg aux jours 8 et 15 du premier mois de traitement.
Au cours d’un essai de phase II les patients qui ont subi une résection complète de la tumeur osseuse à cellules géantes ont reçu le traitement pendant 6 mois supplémentaires après la chirurgie conformément au protocole.
Les patients atteints de tumeurs osseuses à cellules géantes doivent être examinés à intervalles réguliers pour déterminer si le traitement leur est toujours bénéfique. Chez les patients dont la maladie est contrôlée par XGEVA, l’effet de l’interruption ou de l’arrêt du traitement n’a pas été évalué, cependant des données limitées chez ces patients n’indiquent pas de rechute de la maladie à l’arrêt du traitement.
Insuffisance rénale
Aucune adaptation de la posologie n’est nécessaire chez les patients atteints d’insuffisance rénale (voir les rubriques 4.4 pour les recommandations relatives à la surveillance de la calcémie, 4.8 et 5.2).
Insuffisance hépatique
La sécurité et l’efficacité du denosumab n’ont pas été étudiées chez les patients présentant une insuffisance hépatique (voir rubrique 5.2).
Patients âgés (âge ≥ 65 ans)
Aucune adaptation de la posologie n’est nécessaire chez les patients âgés (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de XGEVA n’ont pas été établies dans la population pédiatrique (âge < 18 ans), excepté chez les adolescents (âgés de 12 à 17 ans) à maturité squelettique atteints de tumeurs osseuses à cellules géantes.
XGEVA n’est pas recommandé chez l’enfant (âge < 18 ans), excepté chez les adolescents (âgés de 12 à 17 ans) à maturité squelettique atteints de tumeurs osseuses à cellules géantes (voir rubrique 4.4).
Traitement des adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes non résécables ou pour lesquels la résection chirurgicale est susceptible d’entraîner une morbidité sévère : la posologie est la même que chez l’adulte.
Chez l’animal, l’inhibition de RANK/RANK‑ligand (RANKL) a été associée à une inhibition de la croissance osseuse et à une absence de poussée dentaire. Ces modifications ont été partiellement réversibles à l’arrêt de l’inhibition de RANKL (voir rubrique 5.3).
Mode d’administration
Injection sous‑cutanée.
XGEVA 120 mg/1,7 mL solution en flacon à usage unique :
L’administration du flacon de 120 mg/1,7 mL ne doit être effectuée que par un professionnel de santé.
XGEVA 120 mg/1,0 mL solution en seringue préremplie :
L’administration à l’aide de la seringue préremplie de 120 mg/1,0 mL peut être effectuée par le patient ou un aidant qui a été formé aux techniques d’injection par un professionnel de santé. La première auto‑administration à l’aide de la seringue préremplie XGEVA doit être supervisée par un professionnel de santé.
Pour les instructions concernant l’utilisation, la manipulation et l’élimination, voir rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Hypocalcémie sévère non traitée (voir rubrique 4.4).
Lésions non cicatrisées résultant d’une chirurgie bucco‑dentaire.
4.8 Effets indésirables
Résumé du profil de sécurité
Le profil général de sécurité de XGEVA est cohérent dans toutes les indications approuvées.
Une hypocalcémie a été très fréquemment rapportée suite à l’administration de XGEVA, le plus souvent au cours des deux premières semaines suivant l’injection. L’hypocalcémie peut être sévère et symptomatique (voir rubrique 4.8 ‑ Description d’effets indésirables sélectionnés). Les diminutions du calcium sérique étaient généralement gérées de manière appropriée par une supplémentation en calcium et en vitamine D. Les effets indésirables les plus fréquents associés à XGEVA sont les douleurs musculo‑squelettiques. Des cas d’ostéonécrose de la mâchoire (voir rubriques 4.4 et 4.8 ‑ description d’effets indésirables sélectionnés) ont été fréquemment observés chez les patients traités par XGEVA.
Tableau récapitulatif des effets indésirables
La convention suivante a été utilisée pour la classification des effets indésirables basée sur les taux d’incidence au cours de quatre études cliniques de phase III, de deux études de phase II et de l’expérience après commercialisation (voir tableau 1) : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence et de classe de système d’organe, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1. Effets indésirables rapportés chez des patients présentant une affection maligne avancée avec atteinte osseuse, un myélome multiple ou une tumeur osseuse à cellules géantes
Classe MedDRA de système d’organe | Catégorie de fréquence | Effet indésirable |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) | Fréquent | Second cancer primitif1 |
Affections du système immunitaire | Rare | Hypersensibilité médicamenteuse1 |
Rare | Réaction anaphylactique1 | |
Troubles du métabolisme et de la nutrition | Très fréquent | Hypocalcémie1, 2 |
Fréquent | Hypophosphatémie | |
Peu fréquent | Hypercalcémie après l’arrêt du traitement chez les patients atteints de tumeurs osseuses à cellules géantes3 | |
Affections respiratoires, thoraciques et médiastinales | Très fréquent | Dyspnée |
Affections gastro-intestinales | Très fréquent | Diarrhée |
Fréquent | Extraction dentaire | |
Affections de la peau et du tissu sous-cutané | Fréquent | Hyperhidrose |
Peu fréquent | Éruptions lichénoïdes d’origine médicamenteuse1 | |
Affections musculo-squelettiques et du tissu conjonctif | Très fréquent | Douleur musculo‑squelettique1 |
Fréquent | Ostéonécrose de la mâchoire1 | |
Peu fréquent | Fractures fémorales atypiques1 | |
Fréquence indéterminée | Ostéonécrose du conduit auditif externe3,4 |
1 Voir rubrique Description d’effets indésirables sélectionnés
2 Voir rubrique Autres populations particulières
3 Voir rubrique 4.4
4 Effet de classe
Description d’effets indésirables sélectionnés
Hypocalcémie
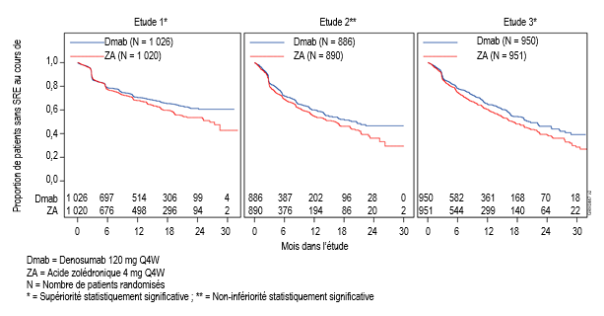
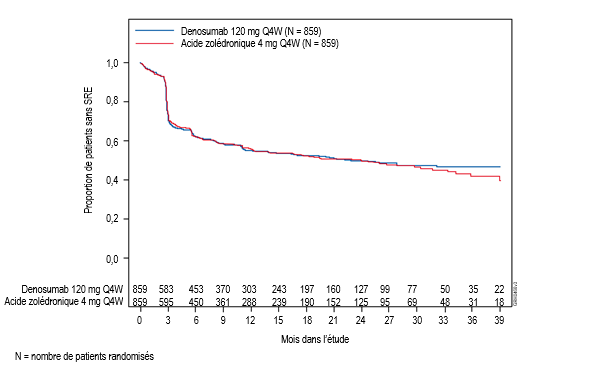
Une incidence plus élevée d’hypocalcémie a été observée chez les patients traités par le denosumab comparé à l’acide zolédronique dans les essais cliniques de prévention des complications osseuses (SRE : skeletal related events).
L’incidence la plus élevée d’hypocalcémie a été observée dans une étude de phase III chez des patients présentant un myélome multiple. Une hypocalcémie a été rapportée chez 16,9 % des patients traités par XGEVA et chez 12,4 % des patients traités par l’acide zolédronique. Une diminution de la calcémie de grade 3 a été observée chez 1,4 % des patients traités par XGEVA et chez 0,6 % des patients traités par l’acide zolédronique. Une diminution de la calcémie de grade 4 a été observée chez 0,4 % des patients traités par XGEVA et chez 0,1 % des patients traités par l’acide zolédronique.
Lors de trois essais cliniques de phase III contrôlés contre comparateur actif menés chez des patients présentant une pathologie maligne avec atteinte osseuse, une hypocalcémie a été rapportée chez 9,6 % des patients traités par XGEVA et chez 5,0 % des patients traités par l’acide zolédronique.
Une diminution de grade 3 de la calcémie a été observée chez 2,5 % des patients traités par XGEVA et chez 1,2 % des patients traités par acide zolédronique. Une diminution de grade 4 de la calcémie a été observée chez 0,6 % des patients traités par XGEVA et chez 0,2 % des patients traités par acide zolédronique (voir rubrique 4.4).
Dans deux essais cliniques de phase II avec un bras unique menés chez des patients atteints de tumeurs osseuses à cellules géantes, une hypocalcémie a été rapportée chez 5,7 % des patients. Aucun des évènements indésirables n’a été considéré comme grave.
Des cas d’hypocalcémie symptomatique sévère (incluant des cas d’issue fatale) ont été rapportés après la commercialisation de XGEVA, la majorité des cas survenant durant les premières semaines d’initiation de la thérapie. Les exemples de manifestations cliniques d’hypocalcémie symptomatique sévère ont inclus allongement de l’intervalle du QT, tétanie, convulsions et altération de l’état mental (y compris coma) (voir rubrique 4.4). Les symptômes d’hypocalcémie survenus au cours des études cliniques incluaient des paresthésies ou raideurs musculaires, contractions, spasmes et crampes musculaires.
Ostéonécrose de la mâchoire (ONM)
Au cours des essais cliniques, l’incidence des ONM a été plus élevée lors des durées d’exposition plus longues ; des ONM ont également été diagnostiquées après l’arrêt du traitement par XGEVA avec une majorité de cas se déclarant dans les 5 mois après la dernière dose. Les patients ayant des antécédents d’ONM ou d’ostéomyélite de la mâchoire, un problème dentaire ou à la mâchoire nécessitant une intervention chirurgicale, une chirurgie bucco‑dentaire non cicatrisée ou toute intervention dentaire invasive planifiée ont été exclus des essais cliniques.
Lors des essais cliniques de prévention des SRE, une incidence plus élevée d’ONM a été observée chez les patients traités par le denosumab comparé à l’acide zolédronique. L’incidence la plus élevée d’ONM a été observée dans une étude de phase III chez des patients présentant un myélome multiple. Durant la phase de traitement en double aveugle de cette étude, les ONM ont été confirmées chez 5,9 % des patients traités par XGEVA (exposition médiane de 19,4 mois ; extrêmes : 1 ‑ 52 mois) et chez 3,2 % des patients traités par l’acide zolédronique. À la fin de la phase de traitement en double aveugle de cette étude, l’incidence ajustée exprimée en patient‑année des ONM confirmées dans le groupe XGEVA (exposition médiane de 19,4 mois ; extrêmes : 1 ‑ 52 mois), était de 2,0 pour 100 patients‑années pendant la première année de traitement, de 5,0 pendant la seconde année et de 4,5 par la suite. Le temps médian d’apparition d’ONM était de 18,7 mois (extrêmes : 1 ‑ 44 mois).
Au cours des trois essais cliniques de phase III contrôlés contre comparateur actif menés chez des patients présentant une affection maligne avec atteinte osseuse, une ONM a été confirmée dans les premières phases de traitement chez 1,8 % des patients traités par XGEVA (exposition médiane de 12,0 mois ; extrêmes : 0,1–40,5 mois) et chez 1,3 % des patients traités par acide zolédronique. Les caractéristiques cliniques de ces cas étaient comparables entre les groupes de traitement. La plupart des patients ayant présenté une ONM confirmée avaient des antécédents d’extraction dentaire, de mauvaise hygiène buccale et/ou d’utilisation d’un appareil dentaire (81 % dans les deux groupes de traitement). La plupart des patients recevaient ou avaient reçu une chimiothérapie.
Les essais chez les patients atteints d’un cancer du sein ou de la prostate comprenaient une phase d’extension de traitement par XGEVA (exposition médiane globale de 14,9 mois ; extrêmes : 0,1 ‑ 67,2 mois). Une ONM a été confirmée chez 6,9 % des patients atteints d’un cancer du sein et d’un cancer de la prostate pendant la phase d’extension du traitement.
L’incidence globale ajustée exprimée en patient‑année des ONM confirmées était de 1,1 pour 100 patients‑années pendant la première année de traitement, de 3,7 pendant la seconde année et de 4,6 par la suite. Le temps médian d’apparition d’ONM était de 20,6 mois (extrêmes : 4 – 53 mois).
Une étude observationnelle, rétrospective, non randomisée menée chez 2 877 patients atteints d’un cancer traités par XGEVA ou acide zolédronique en Suède, au Danemark et en Norvège a montré que le taux d'incidence à 5 ans des ONM confirmées sur le plan médical était de 5,7 % (IC 95 % : 4,4‑7,3 ; durée médiane de suivi de 20 mois [extrêmes : 0,2 ‑ 60]) dans une cohorte de patients recevant XGEVA et de 1,4 % (IC 95 % : 0,8‑2,3 ; durée médiane de suivi de 13 mois [extrêmes : 0,1 ‑ 60]) dans une cohorte de patients distincte recevant de l’acide zolédronique. Le taux d'incidence à cinq ans des ONM chez les patients passant de l’acide zolédronique à XGEVA était de 6,6 % (IC 95 % : 4,2‑10,0 ; durée médiane de suivi de 13 mois [extrêmes : 0,2 ‑ 60]).
Dans une étude de phase III menée chez des patients atteints de cancer de la prostate non métastatique (population de patients dans laquelle XGEVA n’est pas indiqué), avec une durée d’exposition au traitement plus longue allant jusqu’à 7 ans, l’incidence ajustée exprimée en patient‑année des ONM confirmées était de 1,1 pour 100 patients‑années pendant la première année de traitement, de 3,0 pendant la seconde année et de 7,1 par la suite.
Dans une étude clinique de phase II en ouvert, à long terme, menée chez des patients atteints de tumeurs osseuses à cellules géantes (étude 6, voir rubrique 5.1), l’ONM était confirmée chez 6,8 % des patients, dont un adolescent (nombre médian de 34 doses ; extrêmes : 4 ‑ 116). À la fin de l’étude, la durée médiane de l’étude incluant la phase de suivi de sécurité était de 60,9 mois (extrêmes : 0 ‑ 112,6 mois). L’incidence ajustée exprimée en patient‑année des ONM confirmées était globalement de 1,5 pour 100 patients‑années (de 0,2 pour 100 patients‑années pendant la première année de traitement, de 1,5 pendant la seconde année, de 1,8 pendant la troisième année, de 2,1 pendant la quatrième année, de 1,4 pendant la cinquième année et de 2,2 par la suite). Le temps médian d’apparition de l'ONM était de 41 mois (extrêmes : 11 ‑ 96 mois).
L’étude 7 a été menée pour continuer pendant 5 années supplémentaires ou plus le suivi des patients atteints de tumeurs osseuses à cellules géantes qui étaient traités dans l’étude 6. Une ONM a été signalée chez 6 patients (11,8 %) parmi les 51 patients exposés, avec un total médian de 42 doses de denosumab. Trois de ces cas d’ONM ont été confirmés sur le plan médical.
Réactions d’hypersensibilité liées au médicament
Après la mise sur le marché, des réactions d’hypersensibilité, incluant de rares cas de réactions anaphylactiques, ont été rapportées chez des patients recevant XGEVA.
Fractures atypiques du fémur
Globalement, dans le programme d’études cliniques, des fractures fémorales atypiques ont été rapportées peu fréquemment chez les patients traités par XGEVA, et le risque augmentait avec la durée du traitement. Ces événements sont survenus en cours de traitement et jusqu’à 9 mois après l’arrêt du traitement (voir rubrique 4.4).
Dans le programme d’études cliniques sur les tumeurs osseuses à cellules géantes, des fractures fémorales atypiques ont été rapportées fréquemment chez les patients traités par XGEVA. Dans l’étude 6, l’incidence des fractures fémorales atypiques confirmées a été de 0,95 % (5/526) chez les patients atteints de tumeurs osseuses à cellules géantes. Au cours de l’étude 7 de suivi, l’incidence des fractures fémorales atypiques confirmées a été de 3,9 % (2/51) chez les patients exposés au denosumab.
Douleur musculo‑squelettique
Des douleurs musculo‑squelettiques, y compris des cas sévères, ont été signalées chez des patients traités par XGEVA après la commercialisation. Dans les essais cliniques, les douleurs musculo‑squelettiques étaient très fréquentes dans le groupe denosumab et dans le groupe acide zolédronique. Les douleurs musculo‑squelettiques ayant conduit à l’arrêt du traitement étaient peu fréquentes.
Second cancer primitif
Au cours des quatre essais cliniques de phase III contrôlés contre comparateur actif, et menés en double aveugle chez des patients présentant une affection maligne avancée avec atteinte osseuse, des cas de second cancer primitif ont été rapportés chez 1,5 % (54 sur 3 691) des patients traités par XGEVA (exposition médiane de 13,8 mois ; extrêmes : 1,0 ‑ 51,7) et 0,9 % (33 sur 3 688) des patients traités par acide zolédronique (exposition médiane de 12,9 mois ; extrêmes : 1,0 ‑ 50,8).
L’incidence cumulée des cas de second cancer primitif à un an était de 1,1 % pour le denosumab et de 0,6 % pour l’acide zolédronique respectivement.
Aucun profil particulier de type de cancer ou de groupe de cancer relié au traitement n’a été mis en évidence.
Chez les patients atteints d’une tumeur osseuse à cellules géantes, l’incidence de second cancer, y compris de cancers des os ou autres cancers a été de 3,8 % (20/526) dans l’étude 6. Au cours de l’étude 7 de suivi, l’incidence a été de 11,8 % (6/51) chez les patients exposés au denosumab.
Éruptions lichénoïdes d’origine médicamenteuse
Des éruptions lichénoïdes d’origine médicamenteuse (par exemple des réactions de type lichen plan) ont été rapportées chez des patients après commercialisation.
Population pédiatrique
XGEVA a été étudié au cours d’un essai clinique mené en ouvert incluant 28 adolescents à maturité squelettique atteints de tumeurs osseuses à cellules géantes. Sur la base de ces données limitées, le profil des évènements indésirables semble comparable à celui observé chez les adultes.
Des hypercalcémies cliniquement significatives après l’arrêt du traitement ont été rapportées chez des enfants après commercialisation (voir rubrique 4.4).
Autres populations particulières
Insuffisance rénale
Dans une étude clinique menée chez des patients n’étant pas atteints de cancer avancé, insuffisants rénaux sévères (clairance de la créatinine < 30 mL/min) ou dialysés, le risque de développer une hypocalcémie était plus élevé en l’absence de supplémentation en calcium. Le risque de développer une hypocalcémie pendant le traitement par XGEVA augmente avec le degré d’insuffisance rénale. Au cours d’un essai clinique mené chez des patients ne présentant pas de cancer avancé, 19 % des patients présentant une insuffisance rénale sévère (clairance de la créatinine < 30 mL/min) et 63 % des patients dialysés ont développé une hypocalcémie malgré une supplémentation en calcium. L’incidence globale de l’hypocalcémie cliniquement significative était de 9 %.
Une augmentation de l’hormone parathyroïdienne a également été observée chez les patients insuffisants rénaux sévères ou dialysés, traités par XGEVA. La surveillance de la calcémie et un apport adapté de calcium et de vitamine D sont particulièrement importants chez les patients insuffisants rénaux (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance :
Site internet : www.notifieruneffetindesirable.be
e-mail : adr@afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L'AUTORISATION DE MISE SUR LE MARCHÉ
Amgen Europe B.V.
Minervum 7061,
4817 ZK Breda,
Pays‑Bas
8. NUMÉRO(S) D'AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/11/703/001
EU/1/11/703/002
EU/1/11/703/003
EU/1/11/703/004
EU/1/11/703/005
EU/1/11/703/006
10. DATE DE MISE A JOUR DU TEXTE
juillet 2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l'Agence européenne des médicaments http://www.ema.europa.eu/.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 2883296 | XGEVA 120 MG SOL INJECTABLE 1 FL | M05BX04 | € 161,13 | - | Oui | € 12,8 | € 8,5 |
| 2883304 | XGEVA 120 MG SOL INJECTABLE 4 FL | M05BX04 | € 611,62 | - | Oui | € 12,8 | € 8,5 |
| 4792420 | XGEVA 120MG SER PREREMPLIE 4 | € 611,62 | - | Oui | € 12,8 | € 8,5 | |
| 4792438 | XGEVA 120MG SER PREREMPLIE 1 | € 161,13 | - | Oui | € 12,8 | € 8,5 |