1. DÉNOMINATION DU MÉDICAMENT
EVRA 203 microgrammes/24 heures + 33,9 microgrammes/24 heures, dispositif transdermique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque dispositif transdermique de 20 cm2 contient 6 mg de norelgestromine (NGMN) et 600 microgrammes d’éthinylestradiol (EE).
Chaque dispositif transdermique libère une quantité moyenne de 203 microgrammes de NGMN et 33,9 microgrammes d’EE par 24 heures. L’exposition au médicament est décrite avec plus de précisions par le profil pharmacocinétique (voir rubrique 5.2).
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Dispositif transdermique.
Dispositif transdermique mince, de type matriciel, composé de trois couches.
L'inscription « EVRA » est portée sur la face externe beige de la couche de support par tampon à chaud.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Contraception féminine
EVRA est indiqué chez les femmes en âge de concevoir. La sécurité et l’efficacité ont été établies chez des femmes âgées de 18 à 45 ans.
La décision de prescrire EVRA doit être prise en tenant compte des facteurs de risque de la patiente, notamment ses facteurs de risque de thrombo-embolie veineuse (TEV), ainsi que du risque de TEV associé à EVRA en comparaison aux autres CHC (Contraceptifs Hormonaux Combinés) (voir rubriques 4.3 et 4.4).
4.2 Posologie et mode d’administration
Posologie
Afin d’obtenir une efficacité contraceptive maximale, il convient d’informer les patientes de se conformer strictement aux instructions d’utilisation d’EVRA. Pour les instructions de départ, se reporter à la rubrique « comment commencer à utiliser EVRA » ci-dessous.
Un seul dispositif transdermique doit être porté à la fois.
Chaque dispositif transdermique usagé est retiré et immédiatement remplacé par un nouveau à un jour fixe de la semaine (jour de changement), aux 8ème et 15ème jours du cycle. Le changement de dispositif transdermique peut être effectué à tout moment du jour de changement prévu. La quatrième semaine à partir du 22ème jour est un intervalle libre sans dispositif transdermique.
Un nouveau cycle de contraception débute le jour suivant la semaine d’intervalle libre sans dispositif transdermique; le dispositif transdermique suivant d’EVRA doit être appliqué même si aucune hémorragie de privation n’est intervenue ou si l’hémorragie de privation n’est pas encore terminée.
La période sans dispositif transdermique entre deux cycles d’administration ne doit en aucune circonstance dépasser 7 jours. Si cette période sans dispositif transdermique dépasse 7 jours, il est possible que l’utilisatrice ne soit pas protégée contre le risque de grossesse. Un contraceptif non hormonal doit alors être utilisé simultanément pendant 7 jours. Le risque d’ovulation augmente chaque jour au-delà de la période recommandée sans contraceptif. Si un rapport sexuel a eu lieu au cours d’une telle période prolongée sans dispositif transdermique, la possibilité d’une grossesse doit être envisagée.
Populations particulières
Poids corporel supérieur ou égal à 90 kg
L'efficacité contraceptive peut être diminuée chez les femmes pesant 90 kg ou plus.
Insuffisance rénale
EVRA n'a pas été étudié chez les femmes présentant une insuffisance rénale. Aucun ajustement posologique n’est nécessaire, mais comme il est suggéré dans la littérature que la fraction libre de l'éthinylestradiol est plus élevée, une surveillance médicale accrue est nécessaire lors de l’utilisation d’EVRA dans cette population.
Insuffisance hépatique
EVRA n'a pas été étudié chez les femmes présentant une insuffisance hépatique. EVRA est contre-indiqué chez les femmes présentant une insuffisance hépatique (voir rubrique 4.3).
Femmes ménopausées
EVRA n'est pas indiqué chez les femmes ménopausées et n'est pas destiné à être utilisé comme traitement hormonal substitutif.
Population pédiatrique
La sécurité et l'efficacité n'ont pas été établies chez les adolescentes âgées de moins de 18 ans. L’utilisation d'EVRA chez les enfants et adolescentes prépubères n’est pas pertinente.
Mode d’administration
EVRA doit être appliqué sur une peau propre, sèche, saine, intacte et sans pilosité, sur la fesse, l’abdomen, la face extérieure de la partie supérieure du bras ou la partie supérieure du torse, à un endroit où il ne subira aucune friction due à des vêtements serrés. EVRA ne doit pas être placé sur les seins ou sur une peau rouge, irritée ou entaillée. Chaque nouveau dispositif transdermique doit être placé sur la peau à un endroit différent du précédent, afin d’éviter toute irritation potentielle, bien qu’ils puissent être appliqués dans la même région anatomique.
Il convient d’appuyer fermement sur le dispositif transdermique jusqu’à ce que les bordures adhèrent correctement.
Afin d’éviter toute interférence avec les propriétés adhésives du dispositif transdermique, il convient de ne pas appliquer de maquillage, de crèmes, de lotions, de poudres ou autres produits à usage local sur la zone cutanée où le dispositif transdermique est ou sera bientôt mis en place.
Il est recommandé que l’utilisatrice contrôle visuellement son dispositif transdermique chaque jour afin de garantir le maintien d’une adhérence correcte.
Le dispositif transdermique EVRA ne doit pas être coupé, endommagé ou altéré de quelque manière que ce soit car cela peut compromettre l'efficacité contraceptive.
Les dispositifs transdermiques usagés doivent être éliminés avec précautions selon les instructions de la rubrique 6.6.
Comment commencer à utiliser EVRA
Si aucun contraceptif hormonal n’était utilisé au cours du cycle précédent
La contraception avec EVRA débute le 1er jour des règles. Un seul dispositif transdermique est appliqué et porté pendant une semaine complète (7 jours). Le jour de l’application du premier dispositif transdermique (1er jour/Jour du début) détermine le jour de changement des dispositifs transdermiques suivants. Le jour de changement du dispositif transdermique sera le même jour chaque semaine (jours 8, 15, 22 du cycle et jour 1 du cycle suivant). La quatrième semaine est un intervalle libre sans dispositif transdermique à partir du jour 22.
Si le Cycle 1 du traitement débute après le 1er jour du cycle menstruel, un contraceptif non hormonal doit être utilisé simultanément pendant les 7 premiers jours du premier cycle de traitement uniquement.
En relais d’un contraceptif œstroprogestatif oral
Le traitement avec EVRA doit débuter le 1er jour de l’hémorragie de privation. Si aucune hémorragie de privation n’intervient dans les 5 jours qui suivent la prise du dernier comprimé (hormonal) actif, il convient d’éliminer un risque de grossesse avant de commencer le traitement avec EVRA. Si le traitement commence après le premier jour de l’hémorragie de privation, une contraception non hormonale doit être utilisée en parallèle pendant 7 jours.
Si plus de 7 jours s’écoulent après la prise du dernier comprimé actif de contraception orale, la femme peut avoir ovulé et elle doit consulter un médecin avant de commencer un traitement par EVRA. Si un rapport sexuel a eu lieu au cours d’un intervalle prolongé sans pilule, la possibilité d’une grossesse doit être envisagée.
En relais d’une méthode progestative
La femme peut remplacer à tout moment la pilule progestative (ou l’implant le jour de son retrait, ou la méthode injectable au moment de la prochaine injection) mais elle doit associer une méthode de contraception mécanique pendant les 7 premiers jours.
Après un avortement ou une fausse-couche
La femme peut commencer EVRA immédiatement après un avortement ou une fausse-couche intervenant avant la 20ème semaine de gestation. Aucun moyen de contraception supplémentaire n’est nécessaire si EVRA est débuté immédiatement. Notez qu’une ovulation peut intervenir dans les 10 jours qui suivent un avortement ou une fausse-couche.
Après un avortement ou une fausse-couche intervenant durant ou après la 20ème semaine de gestation, EVRA peut être débuté le 21ème jour après l’avortement ou le 1er jour des premières règles spontanées, si celui-ci intervient avant. L’incidence d’une ovulation au 21ème jour après l'avortement (à 20 semaines de gestation) n'est pas connue.
Après un accouchement
Les utilisatrices qui choisissent de ne pas allaiter doivent attendre 4 semaines après l’accouchement pour commencer un traitement contraceptif avec EVRA. Pour les femmes qui commenceraient plus tard, il convient de leur conseiller d'associer une contraception mécanique pendant les 7 premiers jours. Cependant, si un rapport sexuel a déjà eu lieu, il faudra exclure une grossesse avant de pouvoir commencer EVRA ou alors la femme devra attendre son premier cycle menstruel.
Pour les femmes qui allaitent, se reporter à la rubrique 4.6.
Que faire si le dispositif transdermique se décolle entièrement ou partiellement
Si le dispositif transdermique EVRA se décolle complètement ou partiellement et reste décollé, la quantité de médicament administrée est insuffisante.
Si EVRA reste même partiellement décollé :
- pendant moins d’un jour (jusqu’à 24 heures) : il doit être à nouveau appliqué au même endroit ou immédiatement remplacé par un nouveau dispositif transdermique EVRA. Aucun contraceptif supplémentaire n’est nécessaire. Le dispositif transdermique EVRA suivant doit être appliqué le « Jour de changement » habituel.
- pendant plus d’un jour (24 heures ou plus) ou si l’utilisatrice ne sait pas quand le dispositif transdermique s’est soulevé ou décollé : il est possible que l’utilisatrice ne soit pas protégée contre le risque de grossesse. L’utilisatrice doit interrompre le cycle de contraception en cours et entamer immédiatement un nouveau cycle en appliquant un nouveau dispositif transdermique EVRA. Il existe désormais un nouveau « 1er jour » et un nouveau « Jour de changement ». Une contraception non hormonale doit être associée pendant les 7 premiers jours du nouveau cycle uniquement.
Un dispositif transdermique ne doit pas être appliqué à nouveau s’il n’est plus collant ; un nouveau dispositif transdermique doit être appliqué immédiatement. Aucun adhésif ou bandage supplémentaire ne doit être utilisé afin de maintenir le dispositif transdermique EVRA en place.
Si les jours de changement ultérieurs de dispositif transdermique EVRA sont retardés
Au début de tout cycle d’utilisation du dispositif transdermique (Semaine une/1er jour) :
Il est possible que l’utilisatrice ne soit pas protégée contre le risque de grossesse. L’utilisatrice doit appliquer le premier dispositif transdermique du nouveau cycle dès que l’oubli est constaté. Il existe désormais un nouveau « Jour de changement » du dispositif transdermique et un nouveau « 1er jour ». Une contraception non hormonale doit être associée pendant les 7 premiers jours du nouveau cycle. Si un rapport sexuel a eu lieu au cours de cette période prolongée sans dispositif transdermique, la possibilité d’une grossesse doit être envisagée.
Au milieu du cycle (Semaine deux/8ème jour ou Semaine trois/15ème jour) :
- d’un ou deux jours (jusqu’à 48 heures) : l’utilisatrice doit appliquer un nouveau dispositif transdermique EVRA immédiatement. Le dispositif transdermique EVRA suivant doit être appliqué le « Jour de changement » habituel. Si le dispositif transdermique a été porté correctement au cours des 7 jours précédant le premier jour d’oubli, aucun contraceptif supplémentaire n’est nécessaire.
- de plus de deux jours (48 heures ou plus) : il est possible que l’utilisatrice ne soit pas protégée contre le risque de grossesse. L’utilisatrice doit interrompre le cycle de contraception en cours et entamer immédiatement un nouveau cycle de quatre semaines en appliquant un nouveau dispositif transdermique EVRA. Il existe désormais un nouveau « 1er jour » et un nouveau « Jour de changement ». Une contraception non hormonale doit être associée pendant les 7 premiers jours du nouveau cycle.
A la fin du cycle (Semaine quatre/22ème jour)
- Si le dispositif transdermique EVRA n’est pas retiré au début de la semaine 4 (22ème jour), il doit être retiré dès que possible. Le cycle suivant doit débuter le « jour de changement » habituel, c’est-à-dire le lendemain du 28ème jour. Aucun contraceptif supplémentaire n’est nécessaire.
Modification du jour de changement
Afin de retarder d'un cycle la période des règles, la femme doit appliquer un autre dispositif transdermique au début de la 4ème semaine (22ème jour) et donc ne pas observer l’intervalle libre sans dispositif transdermique. Des métrorragies ou des spottings peuvent se produire. Après avoir porté le dispositif transdermique pendant 6 semaines consécutives, un intervalle libre sans dispositif transdermique de 7 jours est nécessaire. Suite à cela, il est possible de reprendre une application régulière d’EVRA.
Si l’utilisatrice souhaite modifier le jour de changement, le cycle en cours doit être achevé et le troisième dispositif transdermique EVRA retiré à la date correcte. Au cours de la semaine sans dispositif transdermique, un nouveau jour de changement peut être sélectionné en appliquant le premier dispositif transdermique EVRA du cycle suivant aussitôt atteint le jour souhaité. L’intervalle libre sans dispositif transdermique ne doit en aucune circonstance dépasser 7 jours consécutifs. Plus l’intervalle libre sans dispositif transdermique est court, plus le risque est élevé pour l'utilisatrice de ne pas avoir d’hémorragies de privation et de présenter des métrorragies ou des spottings pendant le cycle de traitement suivant.
En cas d’irritation cutanée mineure
Si l’utilisation du dispositif transdermique entraîne une irritation gênante, un nouveau dispositif transdermique peut être appliqué à un nouvel endroit jusqu’au jour de changement suivant. Un seul dispositif transdermique doit être porté à la fois.
4.3 Contre-indications
Les contraceptifs hormonaux combinés (CHC) ne doivent pas être utilisés dans les situations suivantes. Si une de ces maladies survient lors de l’utilisation d’EVRA, il faut arrêter EVRA immédiatement.
- Présence ou risque de thrombo-embolie veineuse (TEV)
• Thrombo-embolie veineuse – présence de TEV (patient traité par des anticoagulants) ou antécédents de TEV (p. ex., thrombose veineuse profonde [TVP] ou embolie pulmonaire [EP]) ;
• Prédisposition connue, héréditaire ou acquise, à la thrombo-embolie veineuse, telle qu’une résistance à la protéine C activée (PCa) (y compris une mutation du facteur V de Leiden), un déficit en antithrombine III, un déficit en protéine C, un déficit en protéine S ;
• Intervention chirurgicale majeure avec immobilisation prolongée (voir rubrique 4.4) ;
• Risque élevé de thrombo-embolie veineuse dû à la présence de multiples facteurs de risque (voir rubrique 4.4) ;
- Présence ou risque de thrombo-embolie artérielle (TEA)
• Thrombo-embolie artérielle – présence ou antécédents de thrombo-embolie artérielle (p. ex., infarctus du myocarde [IM]) ou de prodromes (p. ex., angine de poitrine) ;
• Affection cérébrovasculaire – présence ou antécédents d’accident vasculaire cérébral (AVC) ou de prodromes (p. ex., accident ischémique transitoire [AIT]) ;
• Prédisposition connue, héréditaire ou acquise, à la thrombo-embolie artérielle, telle qu’une hyperhomocystéinémie ou la présence d’anticorps anti-phospholipides (anticorps anti-cardiolipine, anticoagulant lupique) ;
• Antécédents de migraine avec symptômes neurologiques focaux ;
• Risque élevé de thrombo-embolie artérielle dû à la présence de multiples facteurs de risque (voir rubrique 4.4) ou d’un facteur de risque sévère tel que :
- diabète avec symptômes vasculaires
- hypertension artérielle sévère
- dyslipoprotéinémie sévère
- Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique 6.1
- Cancer du sein avéré ou suspecté
- Cancer de l’endomètre ou autre néoplasie liée aux œstrogènes avérée ou suspectée
- Anomalies de la fonction hépatique liées à une maladie hépatocellulaire aiguë ou chronique
- Adénomes ou carcinomes hépatiques
- Hémorragie génitale anormale inexpliquée
- Utilisation concomitante avec des médicaments contenant de l’ombitasvir/paritaprévir/ritonavir,dasabuvir, des médicaments contenant du glécaprévir/pibrentasvir ou du sofosbuvir/velpatasvir/voxilaprévir (voir rubrique 4.5).

4.8 Effets indésirables
Résumé du profil de sécurité d’emploi
Les effets indésirables les plus fréquemment rapportés au cours des essais cliniques ont été des céphalées, des nausées, ainsi que des tensions mammaires, apparaissant respectivement chez environ 21,0 %, 16,6 %, et 15,9 % des patientes. Les effets indésirables pouvant survenir en début de traitement mais qui diminuent habituellement après les trois premiers cycles incluent des spottings, des tensions mammaires et des nausées.
Description de certains effets indésirables particuliers
Une augmentation du risque d'événement thrombotique et thrombo-embolique artériel et veineux, incluant l’infarctus du myocarde, l’AVC, les accidents ischémiques transitoires, la thrombose veineuse et l’embolie pulmonaire, a été observée chez les femmes utilisant des CHC ; ceci est abordé plus en détails en rubrique 4.4.
Tableau listant les effets indésirables
La tolérance a été évaluée chez 3 322 femmes sexuellement actives ayant participé à 3 essais cliniques de phase III dont l’objectif était d’évaluer l’efficacité contraceptive. Les patientes ont reçu 6 ou 13 cycles de contraception (EVRA ou un contraceptif oral utilisé comme comparateur), ont pris au moins une dose de médicament et ont fourni des données de tolérance. Le tableau 1 ci-dessous reprend les effets indésirables rapportés au cours des essais cliniques et lors de l’expérience post-marketing. Convention MedDRA pour les fréquences : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare ((≥ 1/10 000 à < 1/1 000)) ; très rare (< 1/10 000) ; indéterminée (ne peut être estimée sur la base des données disponibles).
Table 1: Fréquence des effets indésirables | |
Classe de systèmes organes | Effet indésirable |
Infections et infestations | |
fréquent | Infection fongique (vulvo)-vaginale |
rare | Eruption pustuleuse* |
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) | |
rare | Tumeur hépatique*† |
Affections du système immunitaire | |
peu fréquent | Hypersensibilité |
rare | Réaction anaphylactique* |
Fréquence indéterminée | Exacerbation des symptômes d’angiœdème héréditaire et acquis* |
Trouble du métabolisme et de la nutrition | |
peu fréquent | Hypercholestérolémie |
rare | Hyperglycémie* |
Affections psychiatriques | |
fréquent | Troubles de l’humeur, troubles affectifs et anxiété |
peu fréquent | Insomnie |
rare | Colère* |
Affections du système nerveux | |
très fréquent | Céphalées |
fréquent | Migraine |
rare | Accident vasculaire cérébral**† |
Affections oculaires | |
rare | Intolérance aux lentilles de contact* |
Affections cardiaques | |
rare | Thrombo-embolie artérielle |
Affections vasculaires | |
peu fréquent | Hypertension |
rare | Crise hypertensive* |
Affections respiratoires thoraciques et médiastinales | |
rare | Thrombose (artérielle) pulmonaire*† |
Affections gastro-intestinales | |
très fréquent | Nausées |
fréquent | Douleur abdominale |
rare | Colite* |
Affections hépatobiliaires | |
rare | Cholécystite |
Affections de la peau et du tissu sous-cutané | |
fréquent | Acné |
peu fréquent | Alopécie |
rare | Angioedème* |
Affections musculo-squelettiques et systémiques | |
fréquent | Spasmes musculaires |
Affections des organes de reproduction et du sein | |
très fréquent | Tension mammaire |
fréquent | Dysménorrhée |
peu fréquent | Galactorrhée |
rare | Dysplasie cervicale* |
Troubles généraux et anomalies au site d’administration | |
fréquent | Malaise |
peu fréquent | Œdème généralisé |
rare | Œdème du visage* |
Investigations | |
fréquent | Prise de poids |
peu fréquent | Augmentation de la pression artérielle |
rare | Diminution du taux sanguin de glucose*† |
* Rapporté après commercialisation. | |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir Annexe V.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Gedeon Richter Plc.
Gyömrői út 19-21.
1103 Budapest
Hongrie
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/02/223/001
EU/1/02/223/002
EU/1/02/223/003
10. DATE DE MISE À JOUR DU TEXTE
11/2022.
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments : http://www.ema.europa.eu/.
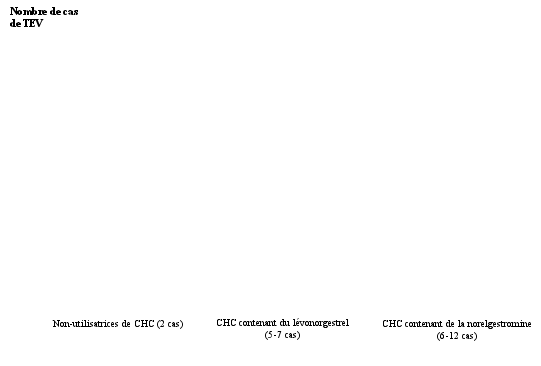
[1] Point central de l’intervalle de 5‑7 pour 10 000 femme-années, sur la base d’un risque relatif, pour les CHC contenant du lévonorgestrel par rapport à leur non-utilisation, d’environ 2,3 à 3,6
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 1777218 | EVRA PATCH 9 | G03AA13 | € 36,43 | - | Oui | € 27,43 | € 27,43 |