RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Ocrevus 920 mg, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 920 mg d’ocrelizumab dans 23 mL (40 mg/mL).
Ocrelizumab est un anticorps monoclonal humanisé, produit dans des cellules d’ovaire de hamster chinois par la technologie de l’ADN recombinant.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable.
Solution limpide à légèrement opalescente, incolore à marron pâle.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Ocrevus est indiqué dans le traitement des patients adultes atteints de formes actives de sclérose en plaques récurrente (SEP-R) définies par des paramètres cliniques ou d’imagerie (voir rubrique 5.1).
Ocrevus est indiqué dans le traitement des patients adultes atteints de sclérose en plaques primaire progressive (SEP-PP) à un stade précoce en termes de durée de la maladie et de niveau du handicap, associé à des données d’imagerie caractéristiques d’une activité inflammatoire (voir rubrique 5.1).
4.2 Posologie et mode d’administration
Le traitement doit être instauré et surveillé par des médecins spécialistes ayant l’expérience du diagnostic et du traitement des affections neurologiques. La première administration doit être effectuée sous surveillance clinique avec un matériel médical approprié nécessaire pour la prise en charge des réactions sévères telles que des réactions sévères liées à l’injection, des réactions d’hypersensibilité et/ou des réactions anaphylactiques (voir rubrique 4.4).
Prémédication pour les réactions liées à l’injection
Les deux prémédications suivantes doivent être administrées peu de temps avant chaque injection d’ocrelizumab afin de réduire le risque de réactions liées à l’injection (RLI) locales et systémiques :
- 20 mg de dexaméthasone par voie orale (ou équivalent)
- un antihistaminique par voie orale (par exemple, desloratadine ou équivalent)
De plus, une prémédication par un antipyrétique (par exemple, paracétamol) peut également être envisagée peu de temps avant chaque administration.
Posologie
La dose recommandée est 920 mg administrée tous les 6 mois.
Aucune division de la dose initiale ou des doses suivantes en administrations séparées n’est nécessaire.
Un intervalle minimal de 5 mois doit être maintenu entre chaque dose d’ocrelizumab.
Injection ou arrêt de traitement en cas de réactions liées à l’injection
Réactions liées à l’injection engageant le pronostic vital
En cas de signes de réactions liées à l’injection engageant le pronostic vital, l’injection doit être immédiatement arrêtée, et le patient doit recevoir un traitement approprié. Le traitement doit être définitivement arrêté chez ces patients (voir rubrique 4.3).
Réactions sévères liées à l’injection
Si un patient présente une réaction sévère liée à l’injection, l’injection doit être immédiatement interrompue, et le patient doit recevoir un traitement symptomatique. L’injection ne sera reprise qu’après la résolution de tous les symptômes (voir rubrique 4.4).
Doses retardées ou oubliées
Si une injection est oubliée, elle doit être administrée dès que possible ; ne pas attendre la dose planifiée suivante. L’intervalle de traitement de 6 mois (avec un minimum de 5 mois) doit être maintenu entre les doses.
Populations particulières
Adultes de plus de 55 ans
Sur la base des données disponibles limitées pour ocrelizumab par voie intraveineuse (voir rubriques 5.1 et 5.2), aucun ajustement posologique n’est nécessaire chez les patients de plus de 55 ans. Les patients ayant participé aux études cliniques continuent de recevoir 600 mg d’ocrelizumab par voie intraveineuse tous les six mois après avoir atteint l’âge de 55 ans. L’utilisation d’ocrelizumab par voie sous-cutanée n’a pas été étudiée chez les patients âgés de plus de 65 ans.
Insuffisance rénale
La sécurité et l’efficacité d’ocrelizumab n’ont pas été formellement étudiées chez les patients présentant une insuffisance rénale. Des patients présentant une insuffisance rénale légère ont été inclus dans les études cliniques. Il n’y a pas d’expérience chez les patients présentant une insuffisance rénale modérée et sévère. Ocrelizumab est un anticorps monoclonal qui est éliminé par catabolisme (c’est-à-dire par dégradation en peptides et en acides aminés), et un ajustement de la dose ne devrait donc pas être nécessaire chez les patients présentant une insuffisance rénale (voir rubrique 5.2).
Insuffisance hépatique
La sécurité et l’efficacité d’ocrelizumab n’ont pas été formellement étudiées chez les patients présentant une insuffisance hépatique. Des patients présentant une insuffisance hépatique légère ont été inclus dans les études cliniques. Il n’y a pas d’expérience chez les patients présentant une insuffisance hépatique modérée et sévère. Ocrelizumab est un anticorps monoclonal qui est éliminé par catabolisme (plutôt que par métabolisme hépatique), et un ajustement de la dose ne devrait donc pas être nécessaire chez les patients présentant une insuffisance hépatique (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité d’ocrelizumab chez les enfants et adolescents âgés de 0 à 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Ocrevus 920 mg, solution injectable n’est pas destiné à une administration intraveineuse et doit être toujours administré par injection sous-cutanée par un professionnel de santé.
Il est important de vérifier les informations présentes sur l’étiquette pour s’assurer que la forme médicamenteuse (intraveineuse ou sous-cutanée) administrée au patient soit bien celle prescrite.
Les patients peuvent commencer le traitement par ocrelizumab par voie intraveineuse ou sous-cutanée et les patients recevant actuellement ocrelizumab par voie intraveineuse peuvent poursuivre le traitement avec ocrelizumab par voie intraveineuse ou passer à Ocrevus 920 mg, solution injectable.
La dose de 920 mg doit être administrée par voie sous-cutanée dans l’abdomen pendant environ 10 minutes. L’utilisation d’un set de perfusion sous-cutanée (par exemple, avec une aiguille à ailettes/papillon) est recommandée. Tout volume résiduel restant dans le set de perfusion sous-cutanée ne doit pas être administré au patient.
Le site d’injection doit être l’abdomen, à l’exception de la zone de 5 cm autour du nombril. Les injections ne doivent jamais être faites dans des zones où la peau est rouge, contusionnée, sensible, ou dure, ou dans des zones où il y a des grains de beauté ou des cicatrices.
Ocrevus solution injectable doit toujours être administré par un professionnel de santé. Pour la première administration, une surveillance après l’injection avec un accès à un matériel médical approprié pour la prise en charge des réactions sévères telles que les RLI, pendant au moins une heure après l’injection est recommandée. Pour les doses suivantes, la nécessité d’une surveillance après l’injection est à l’appréciation du médecin (voir rubrique 4.4).
Pour des instructions sur l’utilisation et la manipulation du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
- Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Infection active en cours (voir rubrique 4.4).
Patients présentant un déficit immunitaire sévère (voir rubrique 4.4).
Affections malignes évolutives connues (voir rubrique 4.4).
4.8 Effets indésirables
Résumé du profil de sécurité
Pendant la période contrôlée des études cliniques pivots, les effets indésirables (EI) les plus importants et les plus fréquemment rapportés ont été les RAP (34,3 % ; 40,1 % dans la SEP-R et la SEP-PP respectivement) et les infections (58,5 % ; 72,2 % dans la SEP-R et la SEP-PP respectivement) (voir rubrique 4.4).
Au total, 2 376 patients ont été inclus durant la période contrôlée des études cliniques pivots ; parmi ces patients, 1 852 ont été inclus dans la phase d’extension en ouvert. Durant la phase d’extension en ouvert, tous les patients sont passés à un traitement par ocrelizumab. 1 155 patients ont terminé la phase d’extension en ouvert, conduisant au total à un recul d’environ 10 ans de traitement continu par ocrelizumab (exposition de 15 515 patients-années) durant la période contrôlée et la phase d’extension en ouvert. Le profil de sécurité global observé pendant la période contrôlée et la phase d’extension en ouvert reste conforme à celui observé durant la période contrôlée.
Le profil de sécurité d’Ocrevus solution injectable a été cohérent avec le profil de sécurité connu d’ocrelizumab par voie intraveineuse présenté dans le tableau 1 ci-dessous à l’exception des effets indésirables très fréquents de RLI.
Liste tabulée des effets indésirables
Les effets indésirables rapportés durant la période contrôlée des études cliniques pivots avec ocrelizumab par voie intraveineuse et issus des notifications spontanées sont listés ci-dessous dans le Tableau 1. Les effets indésirables sont listés par classe de systèmes d’organes MedDRA et par catégories de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles). Au sein de chaque classe de systèmes d’organes, les effets indésirables sont présentés par ordre décroissant de fréquence.
Tableau 1 : Effets indésirables
| Très fréquent | Fréquent | Fréquence indéterminée |
Infections et infestations | Infection des voies respiratoires supérieures, rhinopharyngite, grippe | Sinusite, |
|
Affections hématologiques et du système lymphatique |
| Neutropénie | Neutropénie tardive3 |
Affections respiratoires, thoraciques et médiastinales |
| Toux, |
|
Investigations | Diminution du taux sanguin d’immunoglobulines M | Diminution du taux sanguin d’immunoglobulines G |
|
Lésions, intoxications et complications liées aux procédures | Réactions associées à la perfusion1, réactions liées à l’injection2,3 |
|
|
1 Observé uniquement dans les données poolées d’ocrelizumab par voie intraveineuse.
2 Observé dans une étude en dehors des données poolées d’ocrelizumab par voie intraveineuse (associé à l’administration sous-cutanée).
3 Observé après commercialisation.
Description des effets indésirables sélectionnés
Réactions liées à l’injection (RLI)
Sur la base des symptômes observés, les RLI sont classées en réactions systémiques et réactions locales.
Dans l’étude OCARINA II, 118 patients (naïfs d’ocrelizumab) ont reçu la première injection du produit. Les symptômes les plus fréquents rapportés dans le cadre des RLI systémiques et locales incluaient : des céphalées (2,5 %), des nausées (1,7 %), des érythèmes au site d’injection (29,7 %), des douleurs au site d’injection (14,4 %), des gonflements au site d’injection (8,5 %), et des prurits au site d’injection (6,8 %). Des RLI sont survenues chez 48,3 % de ces patients après la première injection. Sur les 118 patients, 11,0 % et 45,8 % des patients ont présenté respectivement au moins un événement de RLI systémique et de RLI locale. Parmi les patients présentant des RLI, la majorité des patients (82,5 %) a présenté des RLI dans les 24 heures suivant la fin de l’injection, et non pendant l’injection. Toutes les RLI étaient non graves et de sévérité légère (71,9 %) ou modérée (28,1 %). La durée médiane de la RLI a été de 3 jours pour les RLI systémiques et de 4 jours pour les RLI locales. Tous les patients se sont rétablis de leurs RLI, dont 26,3 % ont nécessité un traitement symptomatique.
Dans l’étude OCARINA I, 125 patients ont reçu une ou plusieurs injections sous-cutanée d’ocrelizumab 1200 mg. Sur les 125 patients ayant reçu la première injection, 16,0 % ont présenté au moins un événement de RLI systémique et 64,0 % ont présenté au moins un événement de RLI locale. Sur les 104 patients ayant reçu la seconde injection, l’incidence des RLI systémiques et des RLI locales a diminué respectivement à 7,7 % et 37,5 %. Toutes les RLI étaient non graves et toutes, à l’exception d’une, étaient de sévérité légère ou modérée lors de la première injection. Toutes les RLI étaient non graves et de sévérité légère ou modérée lors de la deuxième injection. 21,2% et 17,9% des patients ayant présenté une RLI ont nécessité respectivement un traitement symptomatique après la première et la deuxième injection.
Ocrelizumab par voie intraveineuse est associé à des réactions associées à la perfusion (RAP), qui peuvent être également dues à la libération de cytokines et/ou d’autres médiateurs chimiques. Les RAP peuvent se manifester par un prurit, une éruption cutanée, une urticaire, un érythème, une irritation de la gorge, une douleur oropharyngée, une dyspnée, un oedème pharyngé ou laryngé, des bouffées vasomotrices, une hypotension, une pyrexie, une fatigue, des céphalées, des vertiges, des nausées, une tachycardie et une anaphylaxie. Des RAP graves, dont certaines nécessitant une hospitalisation, ont été rapportées lors de l’utilisation d’ocrelizumab par voie intraveineuse.
Infection
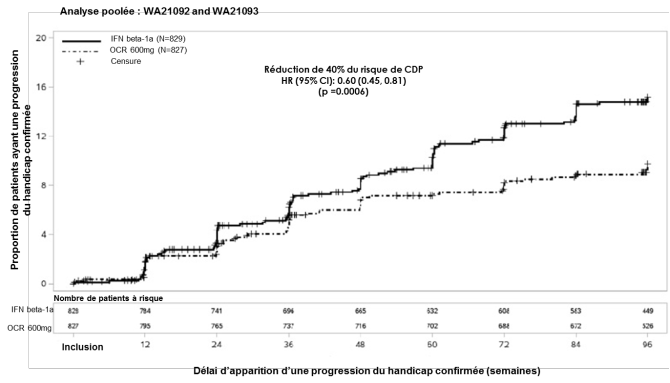
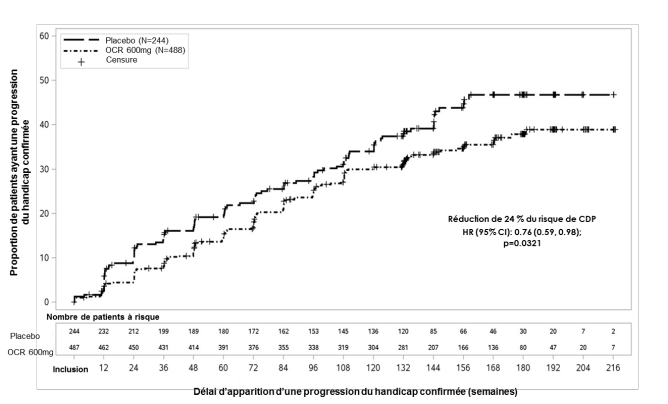
Dans les études contrôlées versus un comparateur actif dans la SEP-R, des infections sont survenues chez 58,5 % des patients ayant reçu ocrelizumab par voie intraveineuse versus 52,5 % des patients ayant reçu l’interféron bêta-1a. Des infections graves sont survenues chez 1,3 % des patients ayant reçu ocrelizumab par voie intraveineuse versus 2,9 % des patients recevant l’interféron bêta-1a. Dans l’étude contrôlée versus placebo dans la SEP-PP, des infections sont survenues chez 72,2 % des patients ayant reçu ocrelizumab par voie intraveineuse versus 69,9 % des patients ayant reçu le placebo. Des infections graves sont survenues chez 6,2 % des patients ayant reçu ocrelizumab par voie intraveineuse versus 6,7 % des patients ayant reçu le placebo.
Tous les patients sont passés à ocrelizumab par voie intraveineuse durant la phase d’extension en ouvert dans les études pivots SEP-R et SEP-PP avec ocrelizumab par voie intraveineuse. Au cours de la phase d’extension en ouvert chez les patients atteints de SEP-R et de SEP-PP, le risque global d'infections graves n'a pas augmenté par rapport à celui observé durant la période contrôlée. Comme observé durant la période contrôlée, le taux d'infections graves chez les patients atteints de SEP-PP est resté plus élevé que celui observé chez les patients atteints de SEP-R.
Conformément à l'analyse précédente relative aux facteurs de risque d'infections graves dans les maladies auto-immunes autres que la SEP (voir rubrique 4.4), une analyse multivariée des facteurs de risque d'infections graves a été conduite sur les données d'exposition cumulée recueillies pendant environ 10 ans à partir de la période contrôlée et la phase d’extension en ouvert des études cliniques pivots. Les facteurs de risque d'infections graves chez les patients atteints de SEP-R comprennent la présence d'au moins une comorbidité, une poussée clinique récente, et un score EDSS (Expanded Disability Status Scale - échelle d’évaluation du handicap) ≥ 6,0. Les facteurs de risque d'infections graves chez les patients atteints de SEP-PP comprennent un indice de masse corporelle supérieur à 25 kg/m2, la présence d'au moins deux comorbidités, un score EDSS ≥ 6,0, et des IgM < limite inférieure de la normale (LIN). Les comorbidités incluaient, sans y être limitées, les affections cardiovasculaires, rénales et des voies urinaires, les infections antérieures, et la dépression.
Infections des voies respiratoires
La proportion d’infections des voies respiratoires a été plus élevée chez les patients traités par ocrelizumab par voie intraveineuse versus l’interféron bêta-1a et versus le placebo.
Dans les études cliniques dans la SEP-R, 39,9 % des patients traités par ocrelizumab par voie intraveineuse et 33,2 % des patients traités par l’interféron bêta-1a ont présenté une infection des voies respiratoires supérieures et 7,5 % des patients traités par ocrelizumab par voie intraveineuse et 5,2 % des patients traités par l’interféron bêta-1a ont présenté une infection des voies respiratoires inférieures.
Dans l’étude clinique dans la SEP-PP, 48,8 % des patients traités par ocrelizumab par voie intraveineuse et 42,7 % des patients ayant reçu le placebo ont présenté une infection des voies respiratoires supérieures, et 9,9 % des patients traités par ocrelizumab par voie intraveineuse et 9,2 % des patients ayant reçu le placebo ont présenté une infection des voies respiratoires inférieures.
Les infections des voies respiratoires rapportées chez les patients traités par ocrelizumab par voie intraveineuse ont essentiellement été légères à modérées (80-90 %).
Herpès
Dans les études cliniques contrôlées versus un comparateur actif (SEP-R), les infections herpétiques ont été rapportées plus fréquemment chez les patients traités par ocrelizumab par voie intraveineuse que chez les patients traités par interféron-bêta-1a, dont zona (2,1 % versus 1,0 %), herpès simplex (0,7 % versus 0,1 %), herpès buccal (3,0 % versus 2,2 %), herpès génital (0,1 % versus 0 %) et infection par le virus de l’herpès (0,1 % versus 0 %). Toutes les infections ont été légères à modérées, à l’exception d’un évènement de grade 3, et les patients ont guéri avec un traitement standard.
Dans l’étude clinique contrôlée versus placebo (SEP-PP), une proportion plus importante de patients avec un herpès buccal (2,7 % versus 0,8 %) a été observée dans le bras traité par ocrelizumab par voie intraveineuse.
Anomalies biologiques
Immunoglobulines
Le traitement par ocrelizumab a entraîné une diminution des immunoglobulines totales au cours de la période contrôlée des études cliniques pivots avec ocrelizumab par voie intraveineuse, principalement due à une réduction des IgM.
Les données des études cliniques issues de la période contrôlée et de la phase d’extension en ouvert des études cliniques pivots ont montré une relation entre la réduction des taux des IgG (et dans une moindre mesure pour les IgM ou les IgA) et l’augmentation du taux d’infections graves. 2,1 % des patients atteints de SEP-R ont présenté une infection grave durant la phase avec des IgG < LIN et 2,3 % des patients atteints de SEP-PP ont présenté une infection grave durant la phase avec des IgG < LIN. La différence de taux d'infections graves entre les patients ayant des IgG < LIN comparé aux patients ayant des IgG ≥ LIN n'a pas augmenté au cours du temps. Le type, la sévérité, la latence, la durée et l’évolution des infections graves observées durant les phases d'immunoglobulines inférieures à la LIN étaient conformes aux infections graves observées chez les patients traités par ocrelizumab pendant la période contrôlée et la phase d’extension en ouvert. Tout au long des 10 années de traitement continu par ocrelizumab, les taux moyens d'IgG chez les patients atteints de SEP-R et SEP-PP sont restés au-dessus de la LIN.
Lymphocytes
Dans la SEP-R, une diminution des lymphocytes < LIN a été observée chez 20,7 % des patients traités par ocrelizumab par voie intraveineuse versus 32,6 % des patients traités par l’interféron bêta-1a. Dans la SEP-PP, une diminution des lymphocytes < LIN a été observée chez 26,3 % des patients traités par ocrelizumab par voie intraveineuse versus 11,7 % des patients traités par placebo.
La majorité de ces diminutions observées chez les patients traités par ocrelizumab par voie intraveineuse étaient de sévérité de grade 1 (entre < LIN et 800 cellules/mm3) et de grade 2 (entre 500 et 800 cellules/mm3). Environ 1 % des patients du groupe ocrelizumab par voie intraveineuse a présenté une lymphopénie de grade 3 (entre 200 et 500 cellules/mm3). Aucun de ces patients n’a présenté une lymphopénie de grade 4 (< 200 cellules/mm3).
Chez les patients traités par ocrelizumab par voie intraveineuse, une augmentation du taux d’infections graves a été observée durant des périodes de diminution confirmée du nombre total de lymphocytes. Le nombre d’infections graves était trop faible pour établir une conclusion définitive.
Neutrophiles
Au cours de la période de traitement contrôlée versus un comparateur actif (SEP-R), une diminution des neutrophiles < LIN a été observée chez 14,7 % des patients traités par ocrelizumab par voie intraveineuse versus 40,9 % des patients traités par l’interféron bêta-1a. Dans l’étude clinique contrôlée versus placebo (SEP-PP), la proportion de patients traités par ocrelizumab par voie intraveineuse présentant une diminution des neutrophiles a été plus élevée (12,9 %) que chez les patients sous placebo (10,0 %) ; parmi ces patients, un pourcentage plus important de patients (4,3 %) dans le groupe ocrelizumab par voie intraveineuse a présenté une neutropénie de grade 2 ou plus versus 1,3 % dans le groupe placebo ; environ 1 % des patients du groupe ocrelizumab par voie intraveineuse a présenté une neutropénie de grade 4 versus 0 % dans le groupe placebo.
La majorité des cas de diminution des neutrophiles a été transitoire (observée une seule fois pour un patient donné traité par ocrelizumab) et de sévérité de Grade 1 (entre < LIN et 1500 cellules/mm3) et 2 (entre 1000 et 1500 cellules/mm3). Globalement, approximativement 1 % des patients dans le groupe ocrelizumab par voie intraveineuse ont eu une neutropénie de Grade 3 ou 4. Un patient avec une neutropénie de Grade 3 (entre 500 et 1000 cellules/mm3) et un patient avec une neutropénie de Grade 4 (< 500 cellules/mm3) ont eu besoin d’un traitement spécifique avec facteur de croissance granulocytaire, et ont continué à recevoir ocrelizumab après résolution de la neutropénie. Une neutropénie peut survenir plusieurs mois après l’administration d’ocrelizumab (voir rubrique 4.4).
Autre
Un patient, qui a reçu 2000 mg d’ocrelizumab par voie intraveineuse, est décédé d’un syndrome de réponse inflammatoire systémique (SIRS) d’étiologie inconnue, suite à un examen IRM 12 semaines après la dernière perfusion ; une réaction anaphylactoïde à l’agent de contraste à base de gadolinium de l’IRM peut avoir contribué à ce SIRS.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1231/003
10. DATE DE MISE À JOUR DU TEXTE
13 février 2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments http://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4841805 | Ocrevus 920 mg, solution injectable | - | € 8941,7 | Oui | - | - |