![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Vyloy 100 mg poudre pour solution à diluer pour perfusion
Vyloy 300 mg poudre pour solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Vyloy 100 mg poudre pour solution à diluer pour perfusion
Chaque flacon de poudre pour solution à diluer pour perfusion contient 100 mg de zolbétuximab.
Vyloy 300 mg poudre pour solution à diluer pour perfusion
Chaque flacon de poudre pour solution à diluer pour perfusion contient 300 mg de zolbétuximab
Après reconstitution, chaque mL de solution contient 20 mg de zolbétuximab.
Le zolbétuximab est produit dans des cellules ovariennes de hamster chinois par la technologie de l’ADN recombinant.
Excipient à effet notoire:
Chaque mL contient 0,21 mg de polysorbate 80.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée blanche à blanc cassé.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
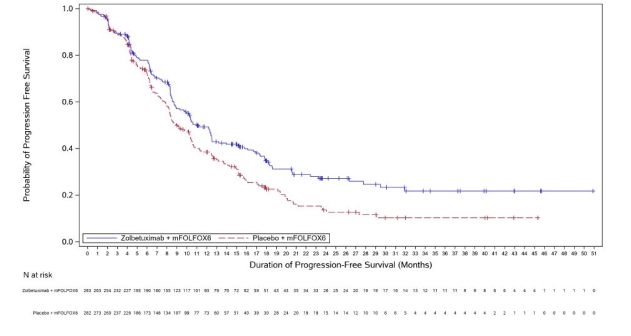
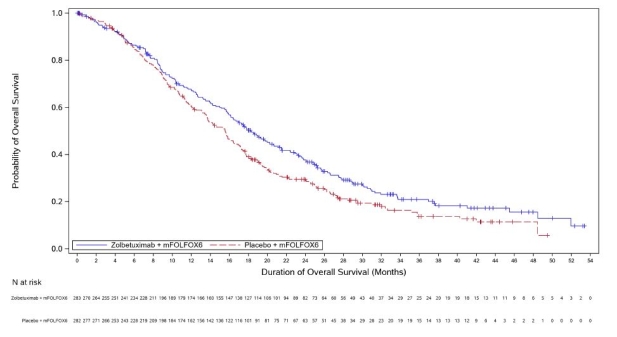
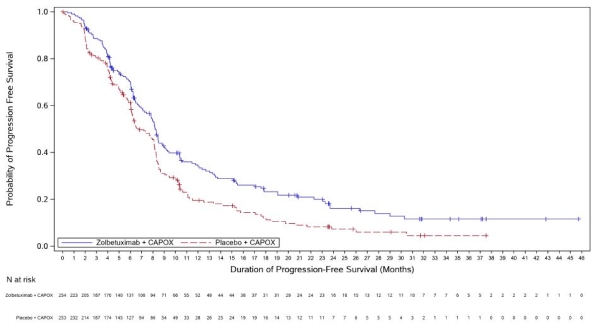
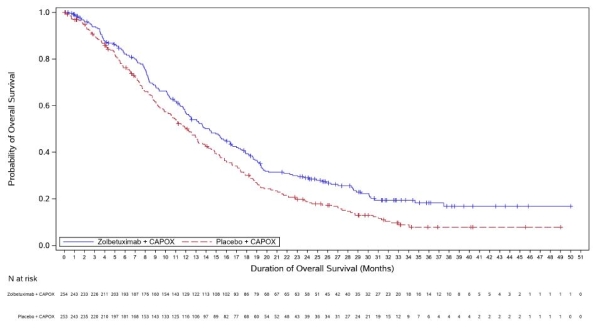
Vyloy, en association à une chimiothérapie combinée à base de fluoropyrimidine et de sels de platine, est indiqué en première ligne de traitement des patients adultes atteints d’un adénocarcinome gastrique ou de la jonction œso-gastrique (JOG) localement avancé non résécable ou métastatique, HER-2 négatif, dont les tumeurs sont Claudine (CLDN) 18.2 positives (voir rubrique 4.2).
4.2 Posologie et mode d’administration
Le traitement doit être prescrit, instauré et supervisé par un médecin expérimenté dans l’utilisation de traitements anticancéreux. Des moyens pour la prise en charge des réactions d’hypersensibilité et/ou des réactions anaphylactiques doivent être disponibles.
Eligibilité des patients
Les patients éligibles doivent présenter une tumeur positive à la protéine CLDN18.2, définie par ≥ 75 % des cellules tumorales présentant une coloration immunohistochimique membranaire CLDN18 modérée à forte, évaluée par test avec marquage CE destiné à cette fin. Si le test avec marquage CE n’est pas disponible, un autre test validé doit être utilisé.
Posologie
Avant l’administration
Si un patient présente des nausées et/ou des vomissements avant l’administration du zolbétuximab, les symptômes doivent revenir à un grade ≤ 1 avant l’administration de la première perfusion.
Avant chaque perfusion de zolbétuximab, les patients doivent recevoir une prémédication associant différents antiémétiques (p. ex., les antagonistes des récepteurs NK-1 et les antagonistes des récepteurs 5-HT3, ainsi que d’autres médicaments selon leurs indications).
Une prémédication associant différents antiémétiques est importante pour la prise en charge des nausées et des vomissements afin d’éviter un arrêt précoce du traitement par zolbétuximab (voir rubrique 4.4). Une prémédication avec des corticostéroïdes systémiques selon les recommandations de traitement locales peut également être envisagée, en particulier avant la première perfusion de zolbétuximab.
Dose recommandée
La dose recommandée doit être calculée en fonction de la surface corporelle (SC) pour la dose de charge et les doses d’entretien de zolbétuximab comme indiqué dans le tableau 1.
Tableau 1. Posologie recommandée du zolbétuximab en fonction de la SC
Dose de charge unique | Doses d’entretien | Durée du traitement |
Le cycle 1, jour 1a, 800 mg/m2 par voie intraveineuse
| Débutant 3 semaines après
| Jusqu’à la progression de la maladie ou la survenue d’une toxicité inacceptable. |
a. La durée du cycle de traitement par zolbétuximab est déterminée en fonction de la chimiothérapie de référence associée (voir rubrique 5.1).
b. Consulter les informations de prescription de la chimiothérapie à base de fluoropyrimidine ou de sels de platine pour les informations relatives à la posologie de la chimiothérapie.
Modifications de dose
Aucune réduction de dose de zolbétuximab n’est recommandée. Les effets indésirables sous zolbétuximab sont pris en charge par une réduction du débit de perfusion, une interruption et/ou un arrêt du traitement comme indiqué au tableau 2.
Tableau 2. Modifications de la dose de zolbétuximab
Effet indésirable | Sévéritéa | Modification de la dose |
Réactions d’hypersensibilité | Réaction anaphylactique, anaphylaxie suspectée, | Interrompre immédiatement la perfusion et arrêter définitivement. |
Grade 2 | Interrompre la perfusion jusqu’à un retour à un grade ≤ 1, puis reprendre à un débit de perfusion réduitb jusqu’à la fin de la perfusion. | |
Réaction liée à la perfusion | Grade 3 ou 4 | Interrompre immédiatement la perfusion et arrêter définitivement. |
Grade 2 | Interrompre la perfusion jusqu’à un retour à un grade ≤ 1, puis reprendre à un débit de perfusion réduitb jusqu’à la fin de la perfusion. | |
Nausée | Grade 2 ou 3 | Interrompre la perfusion jusqu’à un retour à un grade ≤ 1, puis reprendre à un débit de perfusion réduitb jusqu’à la fin de la perfusion. |
Vomissement | Grade 4 | Arrêter définitivement. |
Grade 2 ou 3 | Interrompre la perfusion jusqu’à un retour à un grade ≤ 1, puis reprendre à un débit de perfusion réduitb jusqu’à la fin de la perfusion. |
a. La toxicité a été classifiée selon les critères NCI-CTCAE v4.03 (National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03), le grade 1 indiquant une toxicité légère, le grade 2 une toxicité modérée, le grade 3 une toxicité sévère et le grade 4 une toxicité mettant en jeu le pronostic vital.
b. Le débit de perfusion réduit doit être déterminé selon l’évaluation clinique du médecin en fonction de la tolérance du patient, de la sévérité de la toxicité et du débit de perfusion précédemment toléré (voir rubrique 4.4 pour les recommandations de surveillance des patients).
Populations particulières
Personnes âgées
Aucun ajustement posologique n’est nécessaire chez les patients de ≥ 65 ans (voir rubrique 5.2). Les données des patients de 75 ans et plus ayant été traités par le zolbétuximab sont limitées.
Insuffisance rénale
Aucun ajustement posologique n’est nécessaire chez les patients présentant une atteinte légère (clairance de la créatinine [Clcr] ≥ 60 à < 90 mL/min) ou modérée (Clcr ≥ 30 à < 60 mL/min) de la fonction rénale. Aucune recommandation posologique n’a été établie chez les patients présentant une atteinte sévère de la fonction rénale (Clcr ≥ 15 à < 30 mL/min) (voir rubrique 5.2).
Insuffisance hépatique
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère (bilirubine totale [BT] ≤ limite supérieure de la normale [LSN] et aspartate aminotransférase [ASAT] > LSN, ou BT > 1 à 1,5 × LSN et quel que soit le taux des ASAT). Aucune recommandation posologique n’a été établie chez les patients présentant une insuffisance hépatique modérée (BT > 1,5 à 3 × LSN et quel que soit le taux des ASAT) ou sévère (BT > 3 à 10 × LSN et quel que soit le taux des ASAT) (voir rubrique 5.2).
Population pédiatrique
Il n’existe pas d’utilisation justifiée du zolbétuximab dans la population pédiatrique dans l’indication de l’adénocarcinome gastrique ou de la jonction œso-gastrique.
Mode d’administration
Le zolbétuximab doit être administré par voie intraveineuse. La dose recommandée est administrée par perfusion intraveineuse sur une durée minimum de 2 heures. Le médicament ne doit pas être administré en injection rapide ou en bolus intraveineux.
Si le zolbétuximab et la chimiothérapie à base de fluoropyrimidine et de sels de platine sont administrés le même jour, le zolbétuximab doit être administré en premier.
Pour limiter le risque d’effets indésirables, il est recommandé de commencer chaque perfusion à un débit plus lent pendant 30 à 60 minutes et de l’augmenter progressivement en fonction de la tolérance au cours de la perfusion (voir tableau 3).
Si la durée de la perfusion dépasse la durée recommandée de conservation à température ambiante (≤ 25 °C pendant 8 heures à compter de la fin de la préparation de la solution pour perfusion), la poche de perfusion doit être jetée et une nouvelle poche doit être préparée pour poursuivre la perfusion (voir rubrique 6.3 pour les durées de conservation recommandées).
Tableau 3. Débits de perfusion recommandés pour chaque perfusion de zolbétuximab
Dose de zolbétuximab | Débit de perfusion | ||
Premières 30-60 minutes | Durée restante de la perfusionb | ||
Dose de charge unique (cycle 1, jour 1)a | 800 mg/m2 | 75 mg/m2/h | 150-300 mg/m2/h |
Doses d’entretien | 600 mg/m2 toutes les 3 semaines | 75 mg/m2/h | 150-300 mg/m2/h |
ou | ou | ou | |
400 mg/m2 toutes les 2 semaines | 50 mg/m2/h | 100-200 mg/m2/h | |
a. La durée du cycle de traitement par zolbétuximab est déterminée en fonction de la chimiothérapie de référence associée (voir rubrique 5.1).
b. En l’absence de réactions indésirables au bout de 30-60 minutes, le débit de perfusion peut être augmenté en fonction de la tolérance.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquents de zolbétuximab ont été les nausées (77,2 %), les vomissements (66,9 %), une diminution de l’appétit (42 %), une neutropénie (30,7 %), diminution du nombre de neutrophiles (28,4 %), une perte de poids (21,9 %), de la fièvre (17,4 %), une hypoalbuminémie (17,1 %), un œdème périphérique (13,9 %), une hypertension (9 %), une dyspepsie (7,8 %), des frissons (5,2 %), une hypersialorrhée (3,8 %), des réactions liées à la perfusion (3,2 %) et les hypersensibilités médicamenteuses (1,6 %).
Des effets indésirables graves sont survenus chez 45 % des patients traités par le zolbétuximab. Les effets indésirables graves les plus fréquents ont été les vomissements (6,8 %), les nausées (4,9 %) et une diminution de l’appétit (1,9 %).
Vingt pour cent des patients ont définitivement arrêté de prendre du zolbétuximab en raison d’effets indésirables ; les effets indésirables les plus fréquents entraînant l’arrêt du traitement étaient les vomissements (3,8 %) et les nausées (3,3 %).
Des effets indésirables entraînant une interruption du traitement par le zolbétuximab sont survenus chez 60,9 % des patients ; les effets indésirables les plus fréquents entraînant une interruption du traitement ont été les vomissements (26,6 %), les nausées (25,5 %), la neutropénie (9,8 %), diminution du nombre de neutrophiles (5,9 %), l’hypertension (3,2 %), les frissons (2,2 %), les réactions liées à la perfusion (1,6 %), une diminution de l’appétit (1,6 %) et la dyspepsie (1,1 %).
Tableau récapitulatif des réactions indésirables
Les fréquences des effets indésirables reposent sur deux études de phase 2 et deux études de phase 3 portant sur 631 patients ayant reçu au moins une dose de zolbétuximab de 800 mg/m2 en dose de charge suivie de doses d’entretien de 600 mg/m2 toutes les 3 semaines en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine. Les patients ont été exposés au zolbétuximab pendant une durée médiane de 174 jours (intervalle : 1 à 1 791 jours).
Les effets indésirables observés au cours des études cliniques sont répertoriés dans cette rubrique par catégorie de fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥1/10) ; fréquent (≥1/100, <1/10) ; peu fréquent (≥1/1 000, <1/100) ; rare (≥1/10 000, <1/1 000) ; très rare (<1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les réactions indésirables sont présentées par ordre décroissant de gravité.
Tableau 4. Effets indésirables
Classe de systèmes d’organes MedDRA | Effet indésirable | Catégorie de fréquence |
Affections hématologiques et du système lymphatique | Neutropénie | Très fréquent |
Diminution du nombre de neutrophiles | ||
Affections du système immunitaire | Hypersensibilité médicamenteuse | Fréquent |
Réaction anaphylactique | Peu fréquent | |
Troubles du métabolisme et de la nutrition | Hypoalbuminémie | Très fréquent |
Diminution de l’appétit | ||
Affections vasculaires | Hypertension | Fréquent |
Affections gastro-intestinales | Vomissement | Très fréquent |
Nausée | ||
Dyspepsie | Fréquent | |
Hypersialorrhée | ||
Troubles généraux et anomalies au site d’administration | Fièvre | Très fréquent |
Œdème périphérique | ||
Frissons | Fréquent | |
Investigations | Perte de poids | Très fréquent |
Lésions, intoxications et complications d’interventions | Réaction liée à la perfusion | Fréquent |
Description de certaines réactions indésirables
Réactions d’hypersensibilité
Dans l’analyse des données de sécurité intégrée, des réactions anaphylactiques et des hypersensibilités médicamenteuses de tous grades sont survenues avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine à une fréquence de 0,5 % et 1,6 %, respectivement.
Des cas de réaction anaphylactique et d’hypersensibilité médicamenteuse sévère (grade 3) sont survenus avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine à une fréquence de 0,5 % et de 0,2 %.
Une réaction anaphylactique a entraîné un arrêt définitif du zolbétuximab chez 0,3 % des patients. Une interruption du zolbétuximab a été causée par une hypersensibilité médicamenteuse chez 0,3 % des patients. Le débit de perfusion du zolbétuximab ou de la chimiothérapie à base de fluoropyrimidine et de sels de platine a été réduit chez 0,2 % des patients en raison d’une hypersensibilité médicamenteuse.
Réaction liée à la perfusion
Dans l’analyse des données de sécurité intégrée, des RLP de tous grades sont survenues avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine à une fréquence de 3,2 %.
Une RLP sévère (grade 3) est survenue chez 0,5 % des patients traités avec du zolbétuximab en association à une chimiothérapie à base de fluoropyrimidine et de sels de platine.
Une RLP a entraîné un arrêt définitif du zolbétuximab chez 0,5 % des patients et une interruption du traitement chez 1,6 % des patients. Le débit de perfusion du zolbétuximab ou de la chimiothérapie à base de fluoropyrimidine et de sels de platine a été réduit chez 0,3 % des patients en raison d’une RLP.
Nausée et vomissement
Dans l’analyse des données de sécurité intégrée, des nausées et vomissements de tous grades sont survenus avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine à une fréquence de 77,2 % et 66,9 %, respectivement. Les nausées et les vomissements ont été plus fréquents pendant le premier cycle de traitement, mais leur incidence a diminué au cours des cycles de traitement suivants. Le délai médian d’apparition des nausées et des vomissements était de 1 jour avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine. La durée médiane des nausées et des vomissements était de 3 jours et 1 jour, respectivement, avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine.
Des nausées et des vomissements sévères (grade 3) sont survenus avec le zolbétuximab en association avec une chimiothérapie à base de fluoropyrimidine et de sels de platine à une fréquence de 11,6 % et de 13,6 %.
Des nausées ont entraîné un arrêt définitif du zolbétuximab chez 3,3 % des patients et une interruption du traitement chez 25,5 % des patients. Des vomissements ont entraîné un arrêt définitif du zolbétuximab chez 3,8 % des patients et une interruption du traitement chez 26,6 % des patients. Le débit de perfusion du zolbétuximab ou de la chimiothérapie à base de fluoropyrimidine et de sels de platine a été réduit chez 9,7 % des patients en raison de nausées et chez 7,8 % des patients en raison de vomissements.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration.
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la
pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Astellas Pharma Europe B.V.
Sylviusweg 62
2333 BE Leiden
Pays-Bas
8. NUMÉROS D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/24/1856/001
EU/1/24/1856/002
EU/1/24/1856/003
10. DATE DE MISE À JOUR DU TEXTE
22/01/2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4860649 | VYLOY 100MG PDR SOL DILUER PERFUSION FL 1 | - | - | - | - |