RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Vabysmo 120 mg/mL, solution injectable
Vabysmo 120 mg/mL, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un mL de solution contient 120 mg de faricimab.
Seringue préremplie
Chaque seringue préremplie contient 21 mg de faricimab dans 0,175 mL de solution. Cette quantité est suffisante pour permettre de délivrer une dose unique de 0,05 mL de solution contenant 6 mg de faricimab.
Flacon
Chaque flacon contient 28,8 mg de faricimab dans une solution de 0,24 mL. Cette quantité est suffisante pour permettre de délivrer une dose unique de 0,05 mL de solution contenant 6 mg de faricimab.
Le faricimab est un anticorps humanisé produit en culture dans des cellules mammifères d’ovaires de hamster chinois (CHO) par la technologie de l’ADN recombinant.
Excipient à effet notoire
Chaque solution de 0,05 mL contient 0,02 mg de polysorbate et 0,07 mg de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution injectable (injection)
Solution limpide à opalescente, incolore à jaune brunâtre, avec un pH de 5,5 et une osmolalité de 270 à 370 mOsm/kg.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Vabysmo est indiqué dans le traitement des patients adultes atteints de :
• dégénérescence maculaire liée à l’âge néovasculaire (humide) (DMLAn),
• baisse d’acuité visuelle due à un œdème maculaire diabétique (OMD),
• baisse d’acuité visuelle due à un œdème maculaire secondaire à une occlusion de branche veineuse rétinienne (OBVR) ou de la veine centrale de la rétine (OVCR).
4.2 Posologie et mode d’administration
Ce médicament doit être administré par un médecin qualifié et expérimenté dans les injections intravitréennes.
Posologie
Dégénérescence maculaire liée à l’âge néovasculaire (humide) (DMLAn)
La dose recommandée est de 6 mg (solution de 0,05 mL) administrée par injection intravitréenne toutes les 4 semaines (mensuellement) pour les 3 premières doses.
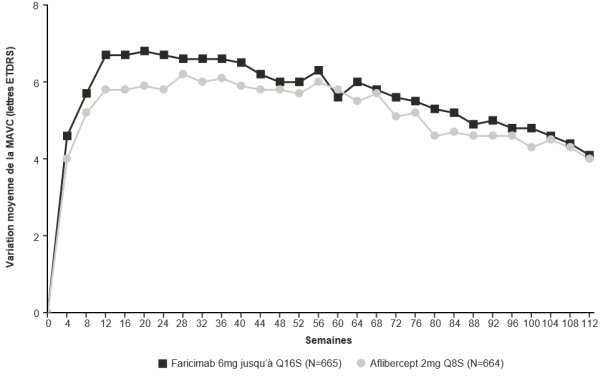
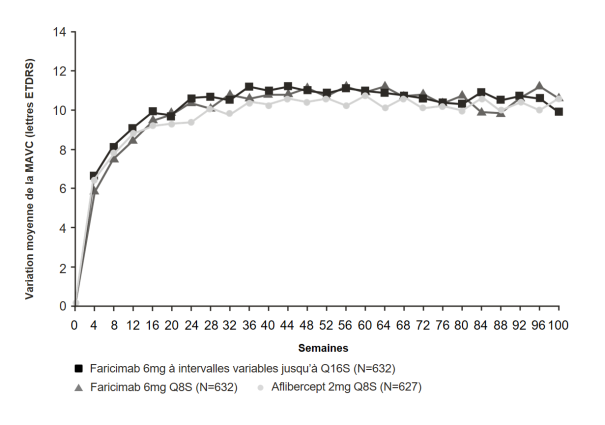
Ensuite, une évaluation de l’activité de la maladie basée sur des résultats anatomiques et/ou visuels est recommandée 16 et/ou 20 semaines après l’initiation du traitement pour que le traitement puisse être individualisé. Chez les patients sans activité de la maladie, l’administration de faricimab toutes les 16 semaines (4 mois) doit être considérée. Chez les patients avec une activité de la maladie, un traitement toutes les 8 semaines (2 mois) ou toutes les 12 semaines (3 mois) doit être considéré. Si les résultats visuels et/ou anatomiques changent, l’intervalle de traitement devra être ajusté en conséquence, et une réduction de l’intervalle doit être mise en œuvre si les résultats visuels et/ou anatomiques se détériorent (voir rubrique 5.1). Les données de sécurité sont limitées concernant les traitements avec des intervalles de 8 semaines ou moins (voir rubrique 4.4). La surveillance entre les visites d’administration doit être programmée en fonction de l’état du patient et du choix du médecin, mais il n’y a pas d’obligation de surveillance mensuelle entre les injections.
Baisse d’acuité visuelle due à un œdème maculaire diabétique (OMD) ou à un œdème maculaire secondaire à une occlusion veineuse rétinienne (OVR)
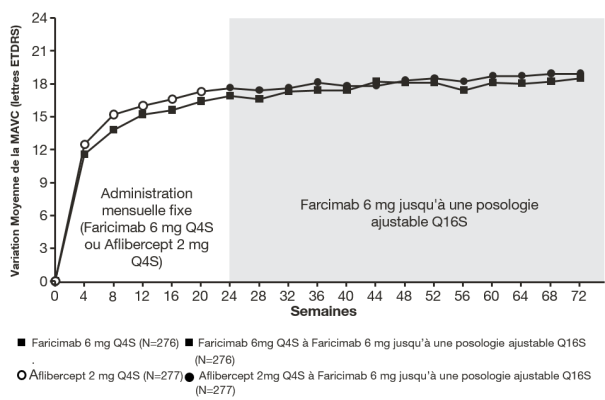
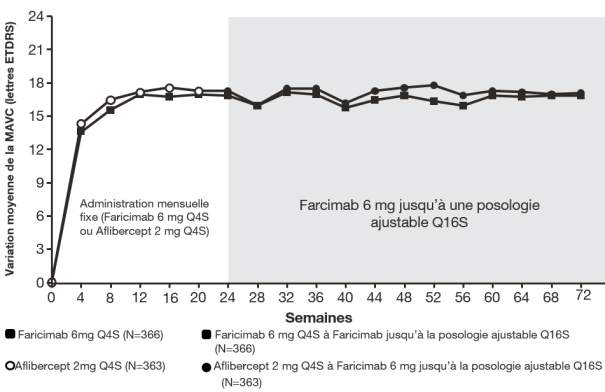
La dose recommandée est de 6 mg (0,05 mL de solution) administrée par injection intravitréenne toutes les 4 semaines (mensuellement) ; 3 injections mensuelles consécutives ou plus peuvent être nécessaires.
Par la suite, le traitement est individualisé en utilisant une approche « treat-and-extend ». Sur avis du médecin en fonction des résultats anatomiques et/ou visuels du patient, l’intervalle des administrations peut être étendu, par palier allant jusqu’à 4 semaines. En cas de détérioration des paramètres visuels et/ou anatomiques, l’intervalle entre deux injections doit être réduit en conséquence (voir rubrique 5.1). Les intervalles de traitement inférieurs à 4 semaines et supérieurs à 4 mois n’ont pas été étudiés.
La surveillance entre les visites d’administration doit être programmée en fonction de l’état du patient et du choix du médecin mais il n’y a pas d’obligation de surveillance mensuelle entre les injections.
Durée du traitement
Ce médicament est destiné à être un traitement à long terme. Si les résultats visuels et/ou anatomiques indiquent que la poursuite du traitement n’est pas bénéfique pour le patient, le traitement doit être arrêté.
Dose retardée ou oubliée
Si une dose est retardée ou oubliée, le patient doit revenir pour être évalué par le médecin lors de la prochaine visite et continuer à être traité, selon le choix du médecin.
Populations particulières
Patients âgés
Aucune adaptation de la posologie n’est nécessaire chez les patients âgés de 65 ans ou plus (voir rubrique 5.2). Les données de sécurité chez les patients atteints de DMLAn, d’OBVR et d’OVCR âgés ≥ 85 ans sont limitées (voir rubrique 4.4).
Insuffisance rénale
Aucune adaptation posologique n’est nécessaire chez les patients atteints d’insuffisance rénale (voir rubrique 5.2).
Insuffisance hépatique
Aucune adaptation posologique n’est nécessaire chez les patients atteints d’insuffisance hépatique (voir rubrique 5.2).
Population pédiatrique
L’utilisation de ce médicament dans la population pédiatrique pour les indications de DMLAn, de l’OMD, de l’OBVR et de l’OVCR n’est pas pertinente.
Mode d’administration
Voie intravitréenne uniquement. Chaque seringue préremplie ou flacon doit être utilisé pour le traitement d’un seul œil.
Vabysmo doit être inspecté visuellement avant l’administration pour vérifier l’absence de particules et de décoloration, auquel cas la seringue préremplie ou le flacon ne doit pas être utilisé.
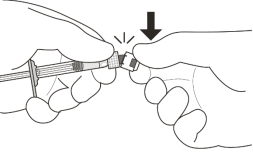
L’injection intravitréenne doit être réalisée dans des conditions aseptiques, incluant la désinfection chirurgicale des mains, l’utilisation d’un champ stérile et d’un spéculum de paupière stérile (ou équivalent). Les antécédents médicaux du patient relatifs aux réactions d’hypersensibilité doivent être attentivement évalués avant d’effectuer l’administration intravitréenne (voir rubrique 4.8). Une anesthésie appropriée et l’application d’un antiseptique local à large spectre pour désinfecter la peau autour de l’œil, la paupière et la surface oculaire doivent être réalisés avant l’injection.
Seringue préremplie
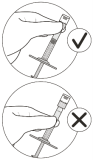
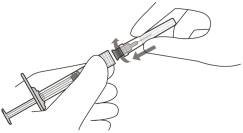
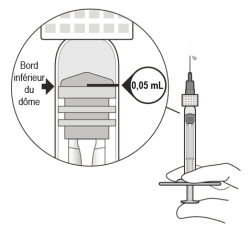
La seringue préremplie contient un excès de volume. Le volume excédentaire doit être éliminé avant d’injecter la dose recommandée. L’injection de la totalité du volume de la seringue préremplie pourrait entraîner un surdosage.
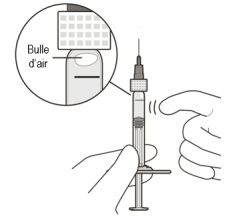
Pour éliminer les bulles d’air en même temps que l’excédent de médicament, pousser lentement le piston jusqu’à ce que le bord inférieur du dôme du bouchon en caoutchouc soit aligné avec la graduation de 0,05 mL (voir rubriques 4.9 et 6.6).
L’aiguille d’injection avec filtre (incluse dans la boîte) doit être insérée 3,5 à 4,0 mm en arrière du limbe dans la cavité vitréenne, en évitant le méridien horizontal et en visant le centre du globe oculaire. Le volume d’injection de 0,05 mL est ensuite administré lentement ; un point d’injection scléral différent doit être utilisé pour les injections ultérieures.
Flacon
L’aiguille d’injection (30 gauge x ½ pouce, non inclus dans la boîte) doit être insérée 3,5 à 4,0 mm en arrière du limbe dans la cavité vitréenne, en évitant le méridien horizontal et en visant le centre du globe oculaire. Le volume d’injection de 0,05 mL est ensuite administré lentement ; un point d’injection scléral différent doit être utilisé pour les injections ultérieures.
Surveillance après l’injection
Après l’injection, tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Immédiatement après l’injection intravitréenne, les patients doivent être surveillés afin de détecter une éventuelle élévation de la pression intraoculaire. Une surveillance appropriée peut consister à une surveillance de la perfusion de la tête du nerf optique ou en réalisant une tonométrie. Si nécessaire, un équipement stérile de paracentèse doit être disponible.
Après l’injection intravitréenne, les patients doivent être informés qu’ils doivent signaler sans délai tout symptôme évocateur d’une endophtalmie (par exemple, perte de vision, douleur oculaire, rougeur de l’œil, photophobie, vision trouble).
Pour les instructions sur la manipulation du médicament avant administration, voir rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Infections oculaires ou périoculaires actives ou suspectées.
Inflammation intraoculaire active.
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés étaient les suivants : cataracte (10 %), hémorragie conjonctivale (7 %), décollement du vitré (4 %), augmentation de la PIO (4 %), corps flottants vitréens (4 %), douleur oculaire (3 %) et déchirure de l’épithélium pigmentaire rétinien (DMLAn uniquement) (3 %).
Les effets indésirables les plus graves étaient l’uvéite (0,5 %), l’endophtalmie (0,4 %), la hyalite (0,4 %), la déchirure de la rétine (0,2 %), le décollement rhegmatogène de la rétine (0,1 %) et la cataracte traumatique (< 0,1 %) (voir rubrique 4.4).
Tableau des effets indésirables
Les effets indésirables rapportés dans les essais cliniques ou pendant la surveillance post-commercialisation sont présentés par classe de systèmes d’organes MedDRA et par fréquence en utilisant la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000) ou fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1 : Fréquences des effets indésirables
Classe de système d’organes MedDRA | Catégorie de fréquence |
Affections oculaires | |
Cataracte | Fréquent |
Hémorragie conjonctivale | Fréquent |
Décollement du vitré | Fréquent |
Augmentation de la pression intraoculaire | Fréquent |
Corps flottants vitréen | Fréquent |
Déchirure de l’épithélium pigmentaire rétinien (DMLAn uniquement) | Fréquent |
Douleur oculaire | Fréquent |
Abrasion de la cornée | Peu fréquent |
Irritation oculaire | Peu fréquent |
Augmentation de la sécrétion lacrymale | Peu fréquent |
Vision trouble | Peu fréquent |
Prurit oculaire | Peu fréquent |
Gêne oculaire | Peu fréquent |
Hyperhémie oculaire | Peu fréquent |
Iritis | Peu fréquent |
Baisse de l’acuité visuelle | Peu fréquent |
Uvéite | Peu fréquent |
Endophtalmie | Peu fréquent |
Sensation de corps étrangers dans l’œil | Peu fréquent |
Hémorragie vitréenne | Peu fréquent |
Hyalite | Peu fréquent |
Iridocyclite | Peu fréquent |
Hyperhémie conjonctivale | Peu fréquent |
Douleur liée à la procédure d’injection | Peu fréquent |
Déchirure de la rétine | Peu fréquent |
Décollement rhegmatogène de la rétine | Peu fréquent |
Baisse de l’acuité visuelle de façon transitoire | Rare |
Cataracte traumatique | Rare |
Vascularite rétinienne* | Indéterminée |
Vascularite rétinienne occlusive* | Indéterminée |
Les termes marqués d’un astérisque (*) sont des effets indésirables qui ont été identifiés sur la base de déclarations spontanées depuis la commercialisation. Etant donné que ces réactions sont rapportées volontairement à partir d’une population de taille incertaine, il n’est pas toujours possible d’estimer de manière fiable leur fréquence.
Description de certains effets indésirables
Vascularite rétinienne ou Vascularite rétinienne occlusive
De rares cas de vascularite rétinienne et/ou de vascularite rétinienne occlusive ont été spontanément rapportés depuis la commercialisation (voir rubrique 4.4). Des cas de vascularite rétinienne et de vascularite rétinienne occlusive ont également été rapportés chez des patients lors de traitements intravitréens.
Effets indésirables liés à la classe de produit
Il existe un risque théorique d’événements thromboemboliques artériels, y compris d’accident vasculaire cérébral et d’infarctus du myocarde, suite à l’utilisation intravitréenne d’inhibiteurs du VEGF. Un faible taux d’incidence d’événements thromboemboliques artériels a été observé au cours des essais cliniques avec le faricimab chez les patients atteints de DMLA, d’OMD, d’OBVR et d’OVCR (voir rubrique 4.4). Dans ces indications, aucune différence notable n’a été observée entre les groupes traités par faricimab et le comparateur.
Immunogénicité
Il existe une possibilité de réponse immunitaire chez les patients traités avec faricimab (voir rubrique 4.4). Après l’administration de faricimab jusqu’à 112 (DMLAn), 100 (OMD) et 72 (OVR) semaines, des anticorps anti-faricimab ont été détectés chez environ 13,8 %, 9,6 % et 14,4 % des patients atteints respectivement de DMLAn, d’OMD et d’OVR randomisés dans le groupe faricimab. La signification clinique des anticorps anti-faricimab sur la sécurité n’est pas claire à ce stade. L’incidence de l’inflammation intraoculaire chez les patients présentant une positivité aux anticorps anti-faricimab était de 12/98 (12,2 % ; DMLAn), 15/128 (11,7 % ; OMD), et 9/95 (9,5% ; OVR), et chez les patients présentant une négativité aux anticorps anti-faricimab était de 8/562 (1,4 % ; DMLAn), 5/1 124 (0,4 % ; OMD), et 10/543 (1,8 % ; OVR). L’incidence des effets indésirables oculaires graves chez les patients présentant une positivité aux anticorps anti-faricimab était de 6/98 (6,1 % ; DMLAn), 14/128 (10,9 % ; OMD), et 7/95 (7,4 % ; OVR) et chez les patients présentant une négativité aux anticorps anti-faricimab était de 23/562 (4,1 % ; DMLAn), 45/1 124 (4,0 % ; OMD), et 34/543 (6,3 % ; OVR). Les anticorps anti-faricimab n’ont pas été associés à un impact sur l’efficacité clinique ou sur la pharmacocinétique systémique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration (voir ci-dessous).
Pour la Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Pour le Luxembourg
Centre Régional de Pharmacovigilance de Nancy
ou Division de la pharmacie et des médicaments
de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Roche Registration GmbH
Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Allemagne
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ)
EU/1/22/1683/001
EU/1/22/1683/002
10. DATE DE MISE À JOUR DU TEXTE
8 mai 2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu/en.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4624631 | Vabysmo 120 mg/ml solution injectable | € 953,6 | - | Oui | - | - |