RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
1. DÉNOMINATION DU MÉDICAMENT
Zejula 100 mg comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient du tosylate de niraparib monohydraté équivalent à 100 mg de niraparib.
Excipients à effet notoire
Chaque comprimé contient 34,7 mg de lactose monohydraté (voir la rubrique 4.4).
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé
Comprimé pelliculé gris, de forme ovale (12 mm × 8 mm) portant l’inscription « 100 mg » sur un côté et portant la mention « Zejula » sur l’autre côté.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Zejula est indiqué :
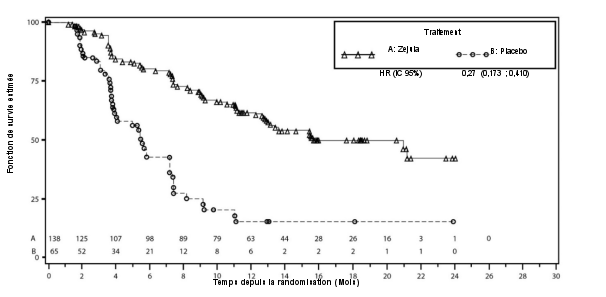
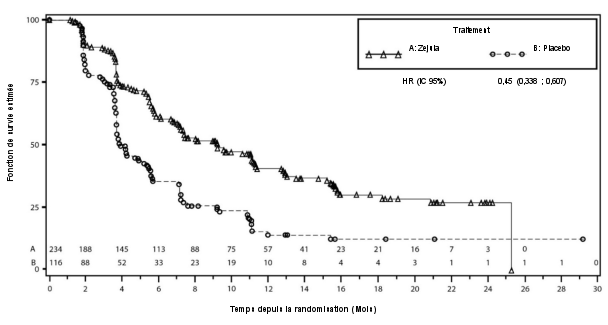
- en monothérapie pour le traitement d’entretien de patientes adultes atteintes d’un cancer épithélial avancé (stades FIGO III et IV) de haut grade de l’ovaire, des trompes de Fallope ou péritonéal primitif, qui sont en réponse (réponse complète ou partielle) à une première ligne de chimiothérapie à base de platine.
- en monothérapie pour le traitement d’entretien de patientes adultes atteintes d’un cancer épithélial séreux de haut grade de l’ovaire, des trompes de Fallope ou péritonéal primitif, sensible au platine et récidivant, qui sont en réponse (réponse complète ou partielle) à une chimiothérapie à base de platine.
4.2 Posologie et mode d’administration
Le traitement par Zejula doit être instauré et supervisé par un médecin expérimenté dans l’utilisation des médicaments anticancéreux.
Posologie
Traitement d’entretien du cancer de l’ovaire en première ligne
La dose initiale recommandée est de 200 mg (deux comprimés à 100 mg), en une prise par jour.
Toutefois, pour les patientes dont le poids est ≥ 77 kg et dont la numération plaquettaire de base est ≥
150,000/μL, la dose initiale recommandée de Zejula est de 300 mg (3 comprimés à 100 mg), en une prise par jour (voir les rubriques 4.4 et 4.8).
Traitement d’entretien du cancer de l’ovaire récidivant
La dose est de trois comprimés de 100 mg une fois par jour, soit l’équivalent d’une dose quotidienne totale de 300 mg
Les patientes doivent être encouragées à prendre leur dose à peu près à la même heure chaque jour. L’administration au coucher est une méthode possible pour gérer les nausées.
Il est recommandé de poursuivre le traitement jusqu’à progression de la maladie ou toxicité.
’Oubli d'une dose
Si les patientes oublient de prendre une dose, elles doivent prendre la dose suivante au moment normalement prévu.
Ajustements posologiques en cas d’effets indésirables
Les ajustements posologiques recommandés en cas d’effets indésirables sont listés dans les
tableaux 1, 2 et 3.
En général, il est recommandé dans un premier temps d’interrompre le traitement (mais pas plus de 28 jours consécutifs) pour permettre à la patiente de récupérer de l’effet indésirable, puis de redémarrer à la même dose. Si l’effet indésirable est récurrent, il est recommandé d’interrompre le
traitement et de reprendre à une dose plus faible. Si les effets indésirables persistent au-delà de 28 jours après l’interruption du traitement, il est recommandé d’arrêter Zejula. Si les effets indésirables ne peuvent pas être contrôlés avec cette stratégie d’interruption et de réduction de dose, il est recommandé d’arrêter Zejula.
Tableau 1: Ajustements posologiques recommandés en cas d’effets indésirables
Niveau de dose initiale | 200 mg | 300 mg |
Première réduction de dose | 100 mg/jour | 200 mg/jour (deux comprimés à 100 mg) |
Seconde réduction de dose | Arrêter Zejula | 100 mg/joura (un comprimé à 100 mg) |
a Si une réduction de dose en dessous de 100 mg/jour est nécessaire, arrêter Zejula.
Tableau 2 : Ajustements posologiques en cas d’effets indésirables non hématologiques
Effet indésirable non hématologique lié au traitement de grade ≥ 3 selon les critères du CTCAE pour lequel une prophylaxie n’est pas considérée comme possible ou effet indésirable qui persiste malgré le traitement | Première survenue : |
Deuxième survenue : | |
Effet indésirable lié au traitement de grade ≥ 3 selon les critères du CTCAE durant plus de 28 jours pendant que la patiente reçoit Zejula à 100 mg/jour | Arrêter le traitement. |
CTCAE=Critères de terminologie communs pour les événements indésirables (« Common Terminology Criteria for Adverse Event »)
Tableau 3 : Ajustements posologiques en cas d’effets indésirables hématologiques
Des effets indésirables hématologiques ont été observés pendant le traitement avec Zejula en particulier pendant la phase initiale du traitement. Il est donc recommandé de surveiller la numération formule sanguine (NFS) chaque semaine pendant le premier mois de traitement et de modifier la dose au besoin. Après le premier mois, il est recommandé de surveiller la NFS mensuellement et périodiquement après ce délai (voir rubrique 4.4). Sur la base des valeurs de laboratoire individuelles, un suivi hebdomadaire durant le deuxième mois peut être justifié. | |
Effet indésirable hématologique nécessitant une transfusion ou un support par facteur de croissance hématopoïétique | • Pour les patientes présentant une numération plaquettaire ≤ 10 000/μL, une transfusion de plaquettes doit être envisagée. S’il existe d’autres facteurs de risque de saignements tels que la co-administration d’anticoagulants ou d’antiplaquettaires, envisager l’interruption de ces médicaments et/ou la transfusion lorsque le nombre de plaquettes est plus élevé. |
Numération plaquettaire < 100 000/μL | Première survenue : |
Deuxième survenue : | |
Neutrophiles < 1 000/µL ou hémoglobine < 8 g/dL | • Suspendre Zejula pendant un maximum de 28 jours et surveiller la numération sanguine une fois par semaine jusqu’à ce que la numération des neutrophiles redevienne ≥ 1 500/μL ou que l’hémoglobine redevienne ≥ 9 g/dL. |
Diagnostic confirmé de syndrome myélodysplasique (SMD) ou de leucémie aiguë myéloïde (LAM) | • Arrêter Zejula de façon permanente. |
Patientes de faible poids corporel en traitement d’entretien du cancer de l’ovaire récidivant
Environ 25 % des patientes de l’étude NOVA pesaient moins de 58 kg et environ 25 % des patientes pesaient plus de 77 kg. L’incidence des effets indésirables de grade 3 ou 4 était plus élevée chez les patientes de faible poids corporel (78 %) que chez celles présentant un poids élevé (53 %). Seulement 13 % des patientes de faible poids corporel sont restées à la dose de 300 mg au-delà du Cycle 3. Une dose initiale de 200 mg pour les patientes pesant moins de 58 kg peut être envisagée.
Patientes âgées
Aucun ajustement posologique n’est nécessaire chez les patientes âgées (≥ 65 ans). Les données cliniques sont limitées chez les patientes âgées de 75 ans ou plus.
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez les patientes atteintes d'insuffisance rénale légère ou modérée. Il n’y a pas de données chez les patientes atteintes d’insuffisance rénale sévère ou d’insuffisance rénale terminale sous hémodialyse ; à utiliser avec prudence chez ces patientes (voir la rubrique 5.2).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire chez les patientes atteintes d'insuffisance hépatique légère (aussi bien lorsque le taux d’aspartate aminotransférase (ASAT) est > à la limite supérieure de la normale (LSN) et le taux de bilirubine totale (BT) est ≤ LSN ou si les taux d’ASAT ou de BT sont > 1,0 x – 1,5 x LSN. Pour les patientes atteintes d'insuffisance hépatique modérée (taux d’ASAT ou de BT>–1,5 x - 3 x LSN), la dose initiale recommandée est de 200 mg une fois par jour. Il n’y a pas de données chez les patientes atteintes d’insuffisance hépatique sévère (taux d’ASAT ou de BT > 3 x LSN) ; à utiliser avec prudence chez ces patientes (voir les rubriques 4.4 et 5.2).
Patientes présentant un indice de performance de 2 à 4 selon l’échelle du statut de performance (ECOG)
Aucune donnée clinique n’est disponible chez les patientes présentant un indice de performance de l’ECOG de 2 à 4.
Population pédiatrique
La sécurité et l’efficacité de Zejula chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. En dehors de ses indications autorisées, Zejula en association avec le dostarlimab a été étudié chez des enfants âgés de 5 ans à moins de 18 ans atteints de tumeurs solides récidivantes ou réfractaires, dont l’ostéosarcome et le neuroblastome ; toutefois, les données issues de l’étude étaient limitées et les résultats de celle-ci n’ont pas permis de conclure que les bénéfices de cette utilisation sont supérieurs aux risques. Les données actuellement disponibles sont décrites dans les rubriques 5.1 et 5.2.
Mode d’administration
Zejula se prend par voie orale.
Il est conseillé de prendre Zejula comprimés en dehors des repas (au moins 1 heure avant ou 2 heures après un repas) ou avec un repas léger (voir rubrique 5.2).
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Allaitement (voir la rubrique 4.6).
4.8 Effets indésirables
Résumé du profil de sécurité
Les effets indésirables de tous grades étant survenus chez ≥ 10 % des 851 patientes traitées par Zejula en monothérapie dans les études PRIMA (à une dose initiale de 200 mg ou de 300 mg ) et NOVA poolées étaient : nausées, anémie, thrombopénie, fatigue, constipation, vomissements, céphalée, insomnie, diminution du nombre de plaquettes, neutropénie, douleur abdominale, appétit diminué, diarrhée, dyspnée, hypertension, asthénie, sensation vertigineuse, diminution du nombre de neutrophiles, toux, arthralgie, dorsalgie, diminution du nombre de globules blancs et bouffée de chaleur.
Les effets indésirables graves les plus fréquents > 1 % (fréquence des effets apparus sous traitement) ont été : thrombopénie et anémie.
Liste tabulée des effets indésirables
Les effets indésirables suivants ont été identifiés sur la base des études cliniques et de la surveillance post-commercialisation chez des patientes traitées par Zejula en monothérapie (voir le tableau 4).
Les fréquences de survenue des effets indésirables sont basées sur les données poolées issues des études cliniques PRIMA et NOVA (à une dose fixe initiale de 300mg/jour), où l’exposition des patientes est connue et sont définies de la manière suivante :
Très fréquent : ≥ 1/10
Fréquent : ≥ 1/100 à < 1/10
Peu fréquent : ≥ 1/1 000 à < 1/100
Rare : ≥ 1/10 000 à < 1/1 000
Très rare : < 1/10 000
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 4 : Liste tabulée des effets indésirables :
Classe de système d’organe | Fréquence de tous grades CTCAE | Fréquence de grade 3 ou 4 CTCAE |
Infections et infestations | Très fréquent | Peu fréquent |
Tumeurs bénignes, malignes et non précisées (y compris kystes et polypes) | Fréquent | Fréquent |
Affections hématologiques et du système lymphatique | Très fréquent | Très fréquent |
Affections du système immunitaire | Fréquent | Peu fréquent |
Troubles du métabolisme et de la nutrition | Très fréquent | Fréquent |
Affections psychiatriques | Très fréquent | Peu fréquent |
Affections du système nerveux | Très fréquent | Peu fréquent |
Affections cardiaques | Très fréquent |
|
Affections vasculaires | Très fréquent | Fréquent |
Affections respiratoires, thoraciques et médiastinales | Très fréquent | Peu fréquent |
Affections gastro-intestinales | Très fréquent | Fréquent |
Affections de la peau et du tissu sous-cutané | Fréquent | Peu fréquent |
Affections musculo-squelettiques et systémiques | Très fréquent | Peu fréquent |
Troubles généraux et anomalies au site d'administration | Très fréquent | Fréquent |
Investigations | Fréquent | Fréquent |
CTCAE=Critères de terminologie communs pour les événements indésirables version 4.02 (« Common Terminology Criteria for Adverse Event »)
a Sur la base des données des essais cliniques réalisés avec le niraparib. Non limité à l’étude clinique pivotale en monothérapie ENGOT-OV16.
b Inclut hypersensibilité, hypersensibilité médicamenteuse, réaction anaphylactoïde, éruption médicamenteuse, angiœdème et urticaire.
c Inclut altération de la mémoire, difficultés de concentration.
Les effets indésirables observés dans le groupe des patientes ayant reçu Zejula à une dose initiale de 200 mg, sur la base du poids corporel ou du taux de plaquettes de base, avaient une fréquence similaire ou inférieure comparativement au groupe ayant reçu une dose fixe initiale de 300 mg/jour (tableau 4).
Voir ci-dessous pour les informations spécifiques concernant la fréquence des thrombopénie, anémie et neutropénie.
Description des événements indésirables sélectionnés
Les effets indésirables hématologiques (thrombopénie, anémie, neutropénie) incluant les diagnostics cliniques et/ou les résultats de laboratoire sont généralement survenus précocement après le début du traitement par niraparib et leur incidence a diminué avec le temps.
Dans NOVA et PRIMA, les patientes éligibles au traitement par Zejula présentaient, avant le début du traitement, les paramètres hématologiques suivants : polynucléaires neutrophiles (PNN)≥ 1 500 /µL ; plaquettes ≥ 100 000 /µL et hémoglobine ≥ 9 g/ dL (NOVA) et ≥ 10 g/dL (PRIMA). Dans le programme clinique, les effets indésirables hématologiques ont été pris en charge par la surveillance des paramètres biologiques et les modifications de dose (voir la rubrique 4.2).
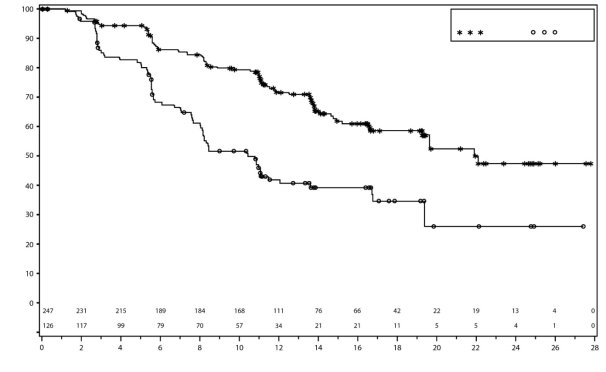
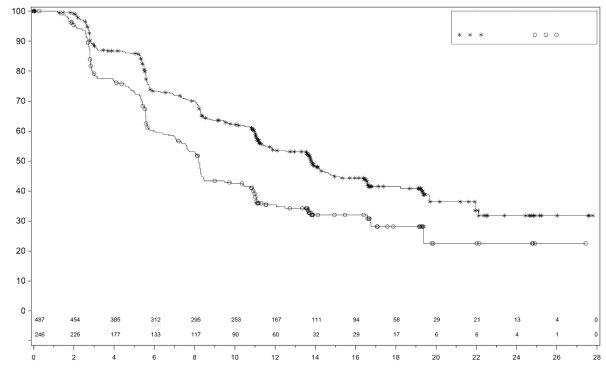
Dans PRIMA, pour les patientes ayant reçu une dose initiale de Zejula en fonction du poids et du taux de base des plaquettes, les thrombopénies, anémies, et neutropénies de grade ≥3 ont diminué respectivement de 48% à 21 %, de 36% à 23 % et de 24% à 15%, comparativement au groupe de patientes ayant reçu une dose fixe d’initiation de 300 mg. Les arrêts de traitement liés à la survenue de thrombopénie, anémie ou neutropénie étaient de 3%, 3% et 2% respectivement.
Thrombopénie
Dans PRIMA, 39 % des patientes recevant Zejula ont présenté une thrombopénie de grade 3/4 comparativement à 0,4 % des patientes dans le groupe placebo, avec un délai médian de 22 jours (intervalle de 15 à 335 jours) entre la première dose et la première survenue de la thrombopénie et avec une durée médiane de 6 jours (intervalle de 1 à 374 jours). Un arrêt de traitement pour cause de thrombopénie était observé chez 4 % des patientes recevant du niraparib.
Dans NOVA, environ 60 % des patientes ont présenté une thrombopénie de tous grades, et 34 % des patientes ont présentées une thrombopénie de grades 3/4. Chez les patientes dont la numération plaquettaire de base était inférieure à 180 × 109/L, une thrombopénie de tout grade et de grade 3/4 s’est produite chez 76 % et 45 % des patientes, respectivement. Le délai médian d’apparition des thrombopénies tous grades confondus et de grade 3/4 était de 22 jours et de 23 jours, respectivement. Le taux de nouveaux cas de thrombopénie après que d’importantes modifications de la dose aient été apportées durant les deux premiers mois du traitement, était de 1,2 % au Cycle 4. La durée médiane des thrombopénies tous grades confondus était de 23 jours et la durée médiane des thrombopénies de grade 3/4 était de 10 jours. Les patientes traitées par Zejula qui développaient une thrombopénie pouvaient avoir un risque accru d’hémorragie. Dans le programme clinique, la thrombopénie a été prise en charge par la surveillance des paramètres biologiques, la modification de la dose et la transfusion de plaquettes, le cas échéant (voir la rubrique 4.2). Des arrêts du traitement dus à des événements de thrombopénie (thrombopénie et numération plaquettaire diminuée) ont eu lieu chez environ 3 % des patientes.
Durant NOVA, 13 % (48/367) des patientes ont présenté un saignement associé à une thrombopénie ; tous les événements hémorragiques survenus de manière concomitante à une thrombopénie présentaient une sévérité de grade 1 ou 2, à l’exception d’un événement de grade 3 associant pétéchie et hématome, observé dans le contexte d’un effet indésirable grave de pancytopénie. Les thrombopénies survenaient plus fréquemment chez les patientes dont la numération plaquettaire de base était inférieure à 180 × 109/L. Environ 76 % des patientes dont la numération plaquettaire de base était < 180 × 109/L et traitées par Zejula ont présenté une thrombopénie de tous grades, et 45 % ont présenté une thrombopénie de grades 3/4. Une pancytopénie a été observée chez < 1 % des patientes recevant du niraparib.
Anémie
Dans PRIMA, 31 % des patientes recevant Zejula ont présenté une anémie de grade 3/4, comparativement à 2 % des patientes dans le groupe placebo, avec un délai médian d’apparition de 80 jours (intervalle 15 à 533 jours) après la première dose et une durée médiane de 7 jours (intervalle 1 à 119 jours). Des arrêts du traitement dus à la survenue d’une anémie ont eu lieu chez 2 % des patientes recevant du niraparib.
Dans NOVA, environ 50 % des patientes recevant Zejula ont présenté une anémie de tous grades, et 25 % des patientes ont présentées une anémie de grades 3/4. Le délai médian d’apparition des anémies tous grades confondus était de 42 jours et de 85 jours pour les événements de grades 3/4. La durée médiane des anémies tous grades confondus était de 63 jours et de 8 jours pour les événements de grades 3/4. Une anémie de tout grade peut persister pendant le traitement avec Zejula. Dans le programme clinique, l’anémie a été prise en charge par la surveillance des paramètres biologiques, la modification de la dose (voir la rubrique 4.2) et, le cas échéant, avec des transfusions de globules rouges. Un arrêt du traitement dû à une anémie a eu lieu chez 1 % des patientes.
Neutropénie
Dans PRIMA, 21 % des patientes recevant Zejula ont présenté une neutropénie de grade 3/4, comparativement à 1 % des patientes dans le groupe placebo, avec un délai médian d’apparition de 29 jours (intervalle de 15 à 421 jours) entre la première dose et la première survenue de la neutropénie et une durée médiane de 8 jours (intervalle de 1 à 42 jours). Des arrêts de traitement dus à la survenue d’une neutropénie ont eu lieu chez 2 % des patientes recevant du niraparib.
Dans NOVA, environ 30 % des patientes ont présenté une neutropénie de tous grades, et 20 % des patientes ont présenté une neutropénie de grade 3/4. Le délai médian d’apparition des neutropénies tous grades confondus était de 27 jours et de 29 jours pour les événements de grade 3/4. La durée médiane des neutropénies tous grades confondus était de 26 jours et de 13 jours pour les événements de grades 3/4. En outre, des facteurs de croissance de la lignée granulocytaire (G-CSF) ont été administrés à environ 6 % des patientes traitées par niraparib comme traitement concomitant pour la neutropénie. Des arrêts de traitement dus à la survenue d’une neutropénie ont eu lieu chez 2 % des patientes.
Syndrome myélodysplasique/Leucémie myéloïde aiguë
Dans les études cliniques, un SMD/LAM est survenu chez 1 % des patientes traitées par Zejula, avec une issue fatale dans 41 % des cas. L'incidence était plus élevée chez les patientes atteintes d'un cancer de l'ovaire en rechute ayant reçu au moins 2 lignes de chimiothérapie à base de platine antérieures et présentant un gBRCAmut après un suivi de survie de 75 mois. Toutes les patientes présentaient des facteurs qui ont potentiellement contribué au développement de SMD/LAM, dans la mesure où elles avaient reçu une chimiothérapie antérieure à base de platine. Nombre d'entre elles avaient également reçu d'autres agents endommageant l'ADN et une radiothérapie. La majorité des cas rapportés concernaient des porteuses du gBRCAmut. Certaines patientes avaient des antécédents de cancer ou de suppression médullaire.
Dans PRIMA, l'incidence des SMD/LAM était de 2,3 % chez les patientes recevant Zejula et de 1,6 % chez les patientes recevant le placebo avec un suivi de 74 mois.
Dans NOVA, menée chez des patientes atteintes d'un cancer de l'ovaire en rechute et ayant reçu au moins deux lignes de chimiothérapie à base de platine, l'incidence globale des SMD/LAM était de 3,8 % chez les patientes recevant Zejula et de 1,7 % chez les patientes recevant le placebo, avec un suivi de 75 mois. Dans les cohortes gBRCAmut et non-gBRCAmut, l'incidence des SMD/LAM était respectivement de 7,4 % et 1,7 % chez les patientes recevant Zejula et de 3,1 % et 0,9 % chez les patientes recevant le placebo.
Hypertension
Dans PRIMA, des cas d’hypertension de grade 3/4, sont survenus chez 6 % des patientes traitées par Zejula comparativement à 1 % des patientes dans le groupe placebo, avec un délai médian d’apparition de 50 jours (intervalle de 1 à 589 jours) après la première dose, et une durée médiane de 12 jours (intervalle 1 à 61 jours). Aucune patiente n’a arrêté Zejula en raison d’une hypertension.
Dans NOVA, une hypertension de tous grades a été observée chez 19,3 % des patientes traitées par Zejula. Une hypertension de grades 3/4 a eu lieu chez 8,2 % des patientes. L’hypertension a été rapidement prise en charge par un traitement à base d’antihypertenseurs. Un arrêt du traitement dû à une hypertension a eu lieu chez < 1 % des patientes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique | Luxembourg |
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
GlaxoSmithKline Trading Services Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Irlande
D24 YK11
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/17/1235/004
EU/1/17/1235/005
EU/1/17/1235/006
EU/1/17/1235/007
10. DATE DE MISE À JOUR DU TEXTE
04/06/2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne du médicament https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4763561 | ZEJULA 100MG COMP PELL PLAQUETTE SECUR. ENFANT 56 | - | € 3800 | Oui | - | - |