RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
▼Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Padcev 20 mg poudre pour solution à diluer pour perfusion
Padcev 30 mg poudre pour solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Padcev 20 mg poudre pour solution à diluer pour perfusion
Un flacon de poudre pour solution à diluer pour perfusion contient 20 mg d'enfortumab vedotin.
Padcev 30 mg poudre pour solution à diluer pour perfusion
Un flacon de poudre pour solution à diluer pour perfusion contient 30 mg d'enfortumab vedotin.
Après reconstitution, chaque mL de solution contient 10 mg d’enfortumab vedotin.
L'enfortumab vedotin est composé d'un anticorps de type IgG1 kappa entièrement humain, conjugué à un agent de perturbation des microtubules, la monométhylauristatine E (MMAE), via un agent de liaison sensible au clivage protéolytique, la maléimidocaproyle valine-citrulline.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée, blanche à blanc cassé.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Tumeur de la vessie infiltrant le muscle (TVIM)
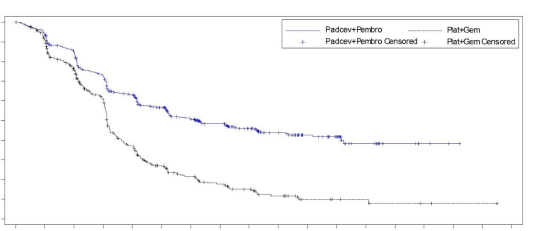
Padcev, en association avec le pembrolizumab, en traitement néoadjuvant puis poursuivi après cystectomie radicale en traitement adjuvant, est indiqué dans le traitement des patients adultes atteints d’une tumeur de la vessie infiltrant le muscle (TVIM) résécable et inéligibles à une chimiothérapie à base de cisplatine.
Carcinome urothélial non résécable ou métastatique
Padcev, en association avec le pembrolizumab, est indiqué dans le traitement de première ligne des patients adultes atteints de carcinome urothélial non résécable ou métastatique et éligibles à une chimiothérapie à base de sels de platine.
Carcinome urothélial localement avancé ou métastatique
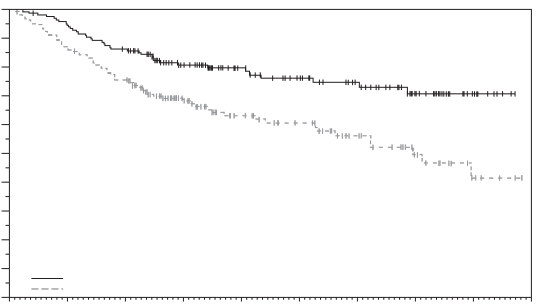
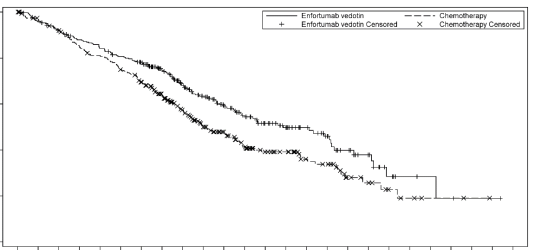
Padcev en monothérapie est indiqué pour le traitement des patients adultes atteints de carcinome urothélial localement avancé ou métastatique, ayant reçu précédemment une chimiothérapie à base de sels de platine et un inhibiteur du récepteur de mort programmée-1 ou un inhibiteur du ligand du récepteur de mort programmée-1 (voir rubrique 5.1).
4.2 Posologie et mode d’administration
Le traitement par Padcev doit être initié et supervisé par un médecin expérimenté dans l'utilisation de traitements anticancéreux. Il convient d’assurer un accès veineux adéquat avant de commencer le traitement (voir rubrique 4.4).
Posologie
Tableau 1. Dose recommandée d’enfortumab vedotin en association avec le pembrolizumab*
Indication | Dose recommandée d’enfortumab vedotin | Durée du traitement |
Traitement néoadjuvant et adjuvant de la tumeur de la vessie infiltrant le muscle (TVIM) | 1,25 mg/kg d’enfortumab vedotin (jusqu’à 125 mg maximum pour les patients ≥ 100 kg) les jours 1 et 8 d’un cycle de 21 jours. | Patients inéligibles au cisplatine |
Carcinome urothélial non résécable ou métastatique | 1,25 mg/kg d’enfortumab vedotin (jusqu’à 125 mg maximum pour les patients ≥ 100 kg) les jours 1 et 8 d’un cycle de 21 jours. | Jusqu’à la progression de la maladie ou la survenue d’une toxicité inacceptable. |
* Les patients doivent recevoir le pembrolizumab après l’enfortumab vedotin lorsque ces deux médicaments sont administrés le même jour. Se référer au RCP du pembrolizumab pour plus d’informations sur la posologie du pembrolizumab.
Tableau 2. Dose recommandée d’enfortumab vedotin en monothérapie
Indication | Dose recommandée d’enfortumab vedotin | Durée du traitement |
Carcinome urothélial localement avancé ou métastatique | 1,25 mg/kg d’enfortumab vedotin (jusqu’à 125 mg maximum pour les patients ≥ 100 kg) les jours 1, 8 et 15 d’un cycle de 28 jours | Jusqu’à la progression de la maladie ou la survenue d’une toxicité inacceptable. |
Tableau 3. Réduction de dose recommandée d’enfortumab vedotin en cas d’effets indésirables | ||
Schéma de réduction de dose | Niveau de dose | |
Dose initiale | 1,25 mg/kg jusqu'à 125 mg | |
Première réduction de dose | 1,0 mg/kg jusqu'à 100 mg | |
Deuxième réduction de dose | 0,75 mg/kg jusqu'à 75 mg | |
Troisième réduction de dose | 0,5 mg/kg jusqu'à 50 mg | |
Modifications de dose
Tableau 4. Interruption, réduction et arrêt de l’administration des doses d’enfortumab vedotin | ||
Effet indésirable | Sévérité* | Modification de dose* |
Réactions cutanées | Suspicion de syndrome de Stevens‑Johnson (SSJ) ou nécrolyse épidermique toxique (NET) ou lésions bulleuses | Suspendre immédiatement le traitement et consulter un spécialiste. |
SSJ ou NET confirmé(e) ; grade 4 ou grade 3 récurrent | Arrêter définitivement. | |
Grade 2 s’aggravant |
| |
Hyperglycémie | Glycémie |
|
Pneumopathie inflammatoire/pneumopathie interstitielle diffuse (PID) | Grade 2 |
|
Grade ≥ 3 | Arrêter définitivement. | |
Neuropathie périphérique | Grade 2 |
|
Grade ≥ 3 | Arrêter définitivement. | |
*La toxicité a été classée selon les Common Terminology Criteria for Adverse Events version 5.0 du National Cancer Institute (Institut national de cancérologie USA) (NCI-CTCAE v5.0), où le grade 1 correspond à léger, le grade 2 à modéré, le grade 3 à sévère et le grade 4 à menaçant la vie du patient. | ||
Populations particulières
Personnes âgées
Aucune adaptation posologique n’est nécessaire chez les patients âgés de 65 ans ou plus (voir rubrique 5.2).
Altération de la fonction rénale
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère [clairance de la créatinine (ClCr) > 60–90 mL/min], modérée (ClCr 30–60 mL/min) ou sévère (ClCr 15–<30 mL/min). L’enfortumab vedotin n’a pas été évalué chez des patients atteints d’insuffisance rénale terminale (ClCr < 15 mL/min) (voir rubrique 5.2).
Altération de la fonction hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une altération légère de la fonction hépatique [bilirubine totale de 1 à 1,5 × limite supérieure de la normale (LSN) et indépendamment du taux d’ASAT, ou bilirubine totale ≤ LSN et ASAT > LSN]. L’enfortumab vedotin a été évalué chez un nombre limité de patients présentant une altération modérée et sévère de la fonction hépatique. La déficience hépatique devrait augmenter l'exposition systémique à la MMAE (la substance cytotoxique), par conséquent, les patients devront être étroitement surveillés pour déceler d’éventuels événements indésirables. En raison de la rareté des données chez les patients atteints de déficience hépatique modérée et sévère, aucune recommandation spécifique de dose ne peut être donnée (voir rubrique 5.2).
Population pédiatrique
L’utilisation d'enfortumab vedotin n’est pas justifiée dans la population pédiatrique pour l’indication de carcinome urothélial localement avancé ou métastatique ni pour celle de TVIM.
Mode d’administration
Padcev doit être administré par voie intraveineuse. La dose recommandée doit être administrée par perfusion intraveineuse pendant 30 minutes. L'enfortumab vedotin ne doit pas être administré en injection rapide ou en bolus intraveineux.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
Enfortumab vedotin en association avec le pembrolizumab
Lorsque l’enfortumab vedotin est administré en association avec le pembrolizumab, se référer au RCP du pembrolizumab avant l’instauration du traitement.
Tumeur de la vessie infiltrant le muscle
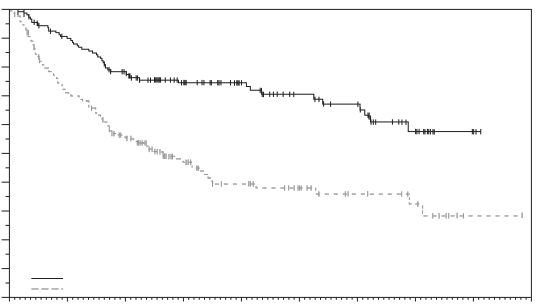
La sécurité de l’enfortumab vedotin a été évaluée en association avec le pembrolizumab chez 167 patients ayant reçu au moins une dose d’enfortumab vedotin de 1,25 mg/kg en association avec le pembrolizumab dans une étude de phase 3 (EV-303) (voir Tableau 5). Les patients ont été exposés à l’enfortumab vedotin en association avec le pembrolizumab pour une durée médiane de 6,3 mois (intervalle : 0,03 à 19,7 mois). La durée médiane d’exposition à l’enfortumab vedotin seul était de 5,5 mois (intervalle : 0,03 à 14,1 mois).
Les effets indésirables les plus fréquents rapportés avec l’enfortumab vedotin en association avec le pembrolizumab ont été les suivants : prurit (47,3 %), alopécie (34,7 %), diarrhée (34,1 %), fatigue (32,3 %), anémie (30,5 %), diminution de l’appétit (28,1 %), dysgueusie (28,1 %), nausées (25,7 %), éruption cutanée (25,1 %), augmentation du taux d’aspartate aminotransférase (24 %), perte de poids (19,8 %), augmentation du taux d’alanine aminotransférase (19,2 %), éruption maculo-papuleuse (16,2 %), sécheresse cutanée (15 %), hypothyroïdie (14,4 %), neuropathie périphérique sensitive (13,8 %), hyperglycémie (12,6 %) et neuropathie périphérique (10,2 %).
L’effet indésirable grave le plus fréquent (≥ 2 %) était la diarrhée (2,4 %). Quarante-et-un pour cent des patients ont arrêté définitivement l’enfortumab vedotin en raison d’effets indésirables ; l’effet indésirable le plus fréquent (≥ 2 %) entraînant un arrêt du traitement était la neuropathie périphérique sensitive (2,4 %).
Des effets indésirables entraînant une interruption du traitement par l’enfortumab vedotin sont survenus chez 44 % des patients. Les effets indésirables les plus fréquents (≥ 2 %) entraînant une interruption du traitement étaient la diarrhée (4,2 %), l’éruption cutanée (4,2 %), la neutropénie (3,6 %), la fatigue (3 %), l’hyperglycémie (3 %) et le prurit (2,4 %).
Des effets indésirables entraînant une réduction de la dose d’enfortumab vedotin sont survenus chez 17 % des patients. Les effets indésirables les plus fréquents (≥ 2 %) entraînant une réduction de la dose étaient l’éruption cutanée (2,4 %) et la perte de poids (2,4 %).
Carcinome urothélial non résécable ou métastatique
La sécurité de l’enfortumab vedotin a été évaluée en association avec le pembrolizumab chez 564 patients ayant reçu au moins une dose d’enfortumab vedotin de 1,25 mg/kg en association avec le pembrolizumab dans une étude de phase 2 (EV-103) et une étude de phase 3 (EV-302) (voir Tableau 5). Les patients ont été exposés à l’enfortumab vedotin en association avec le pembrolizumab pour une durée médiane de 9,4 mois (intervalle : 0,3 à 34,4 mois).
Les effets indésirables les plus fréquents rapportés avec l’enfortumab vedotin en association avec le pembrolizumab ont été les suivants : neuropathie périphérique sensitive (53,4 %), prurit (41,1 %), fatigue (40,4 %), diarrhée (39,2 %), alopécie (38,5 %), éruption maculo-papuleuse (36 %), perte de poids (36 %), diminution de l’appétit (33,9 %), nausées (28,4 %), anémie (25,7 %), dysgueusie (24,3 %), sécheresse cutanée (18,1 %), augmentation du taux d’alanine aminotransférase (16,8 %), hyperglycémie (16,7 %), augmentation du taux d’aspartate aminotransférase (15,4 %), sécheresse oculaire (14,4 %), vomissement (13,3 %), éruption maculeuse (11,3 %), hypothyroïdie (10,5 %) et neutropénie (10,1 %).
Les effets indésirables graves les plus fréquents (≥ 2 %) ont été la diarrhée (3 %) et la pneumopathie inflammatoire (2,3 %). Trente-six pour cent des patients ont arrêté définitivement l’enfortumab vedotin en raison d’effets indésirables ; les effets indésirables les plus fréquents (≥ 2 %) entraînant un arrêt du traitement ont été la neuropathie périphérique sensitive (12,2 %) et l’éruption maculo-papuleuse (2 %).
Des effets indésirables entraînant une interruption du traitement par l’enfortumab vedotin sont survenus chez 72 % des patients. Les effets indésirables les plus fréquents (≥ 2 %) entraînant une interruption du traitement ont été la neuropathie périphérique sensitive (17 %), l’éruption maculo-papuleuse (6,9 %), la diarrhée (4,8 %), la fatigue (3,7 %), la pneumopathie inflammatoire (3,7 %), l’hyperglycémie (3,4 %), la neutropénie (3,2 %), l’augmentation du taux d’alanine aminotransférase (3 %), le prurit (2,3 %) et l’anémie (2 %).
Des effets indésirables entraînant une réduction de la dose d’enfortumab vedotin sont survenus chez 42,4 % des patients. Les effets indésirables les plus fréquents (≥ 2 %) entraînant une réduction de la dose ont été la neuropathie périphérique sensitive (9,9 %), l’éruption maculo-papuleuse (6,4 %), la fatigue (3,2 %), la diarrhée (2,3 %) et la neutropénie (2,1 %).
Enfortumab vedotin en monothérapie
La sécurité de l’enfortumab vedotin en monothérapie a été évaluée chez 793 patients ayant reçu au moins une dose d’enfortumab vedotin de 1,25 mg/kg dans deux études de phase 1 (EV-101 et EV-102), trois études de phase 2 (EV-103, EV-201 et EV-203) et une étude de phase 3 (EV-301) (voir Tableau 5). Les patients étaient exposés à l’enfortumab vedotin pour une durée médiane de 4,7 mois (intervalle : 0,3 à 55,7 mois).
Les effets indésirables les plus fréquents rapportés avec l'enfortumab vedotin ont été les suivants : alopécie (47,7 %), diminution de l’appétit (47,2 %), fatigue (46,8 %), diarrhée (39,1 %), neuropathie périphérique sensitive (38,5 %), nausée (37,8 %), prurit (33,4 %), dysgueusie (30,4 %), anémie (29,1 %), perte de poids (25,2 %), éruption maculo-papuleuse (23,6 %), sécheresse cutanée (21,8 %), vomissement (18,7 %), augmentation du taux d’aspartate aminotransférase (17 %), hyperglycémie (14,9 %), sécheresse oculaire (12,7 %), augmentation du taux d’alanine aminotransférase (12,7 %) et éruption cutanée (11,6 %).
Les effets indésirables graves les plus fréquents (≥ 2 %) ont été la diarrhée (2,1 %) et l'hyperglycémie (2,1 %). Vingt-et-un pour cent des patients ont arrêté définitivement l’enfortumab vedotin en raison d’effets indésirables ; l’effet indésirable le plus fréquent (≥ 2 %) entraînant un arrêt définitif du traitement était la neuropathie périphérique sensitive (4,8 %). Des effets indésirables entraînant une interruption du traitement sont survenus chez 62 % des patients ; les effets indésirables les plus fréquents (≥ 2 %) entraînant une interruption du traitement étaient la neuropathie périphérique sensitive (14,8 %), la fatigue (7,4 %), l’éruption maculo-papuleuse (4 %), l’augmentation du taux d’aspartate aminotransférase (3,4 %), l’augmentation du taux d’alanine aminotransférase (3,2 %), l’anémie (3,2 %), l’hyperglycémie (3,2 %), la diminution du nombre de neutrophiles (3 %), la diarrhée (2,8 %), l’éruption cutanée (2,4 %) et la neuropathie périphérique motrice (2,1 %). Trente-huit pour cent des patients ont présenté un effet indésirable nécessitant une réduction de la dose ; les effets indésirables les plus fréquents (≥ 2 %) entraînant une réduction de la dose étaient la neuropathie périphérique sensitive (10,3 %), la fatigue (5,3 %), l’éruption maculo-papuleuse (4,2 %) et la diminution de l’appétit (2,1 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables observés au cours des études cliniques de l’enfortumab vedotin administré en monothérapie ou en association avec le pembrolizumab ou rapportés lors de l’utilisation post-commercialisation de l’enfortumab vedotin sont répertoriés dans cette rubrique par catégorie de fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 5. Effets indésirables chez les patients traités par l’enfortumab vedotin
Fréquence | Monothérapie | En association avec le pembrolizumab |
Infections et infestations | ||
Fréquent | Sepsis, pneumonie | Sepsis, pneumonie |
Affections hématologiques et du système lymphatique | ||
Très fréquent | Anémie | Anémie |
Fréquent | Thrombocytopénie | Thrombocytopénie |
Fréquence indéterminée1 | Neutropénie, neutropénie fébrile, diminution du nombre de neutrophiles | Neutropénie, neutropénie fébrile, diminution du nombre de neutrophiles |
Affections endocriniennes | ||
Très fréquent |
| Hypothyroïdie |
Troubles du métabolisme et de la nutrition | ||
Très fréquent | Hyperglycémie, diminution de l’appétit | Hyperglycémie, diminution de l’appétit |
Fréquence indéterminée1 | Acidocétose diabétique | Acidocétose diabétique |
Affections du système nerveux | ||
Très fréquent | Neuropathie périphérique sensitive, dysgueusie | Neuropathie périphérique sensitive, dysgueusie |
Fréquent | Neuropathie périphérique, neuropathie périphérique motrice, neuropathie périphérique sensitivo-motrice, paresthésie, hypoesthésie, troubles de la démarche, faiblesse musculaire | Neuropathie périphérique, neuropathie périphérique motrice, neuropathie périphérique sensitivo-motrice, paresthésie, hypoesthésie, troubles de la démarche, faiblesse musculaire, neurotoxicité |
Peu fréquent | Polyneuropathie démyélinisante, polyneuropathie, neurotoxicité, dysfonction motrice, dysesthésie, atrophie musculaire, névralgie, paralysie du nerf sciatique poplité externe, déficit sensoriel, sensation de brûlure de la peau, sensation de brûlure | Dysesthésie, polyneuropathie, myasthénie grave, névralgie, paralysie du nerf sciatique poplité externe, sensation de brûlure de la peau |
Affections oculaires | ||
Très fréquent | Sécheresse oculaire | Sécheresse oculaire |
Affections respiratoires, thoraciques et médiastinales | ||
Fréquent | Pneumopathie inflammatoire/PID2 | Pneumopathie inflammatoire/PID2 |
Affections gastro-intestinales | ||
Très fréquent | Diarrhée, vomissement, nausées | Diarrhée, vomissement, nausées |
Affections de la peau et du tissu sous-cutané | ||
Très fréquent | Alopécie, prurit, éruption cutanée, éruption maculo-papuleuse, sécheresse cutanée | Alopécie, prurit, éruption maculo-papuleuse, sécheresse cutanée |
Fréquent | Éruption d’origine médicamenteuse, exfoliation cutanée, conjonctivite, dermatite bulleuse, cloque, stomatite, syndrome d’érythrodysesthésie palmo-plantaire, eczéma, érythème, éruption érythémateuse, éruption maculeuse, éruption papuleuse, éruption prurigineuse, éruption vésiculeuse | Éruption cutanée, exfoliation cutanée, conjonctivite, dermatite bulleuse, cloque, stomatite, syndrome d’érythrodysesthésie palmo-plantaire, eczéma, érythème, éruption érythémateuse, éruption maculeuse, éruption papuleuse, éruption prurigineuse, éruption vésiculeuse, dermatite |
Peu fréquent | Dermatite exfoliative généralisée, érythème polymorphe, éruption avec exfoliation, pemphigoïde, éruption maculo-vésiculeuse, dermatite, dermatite allergique, dermatite de contact, intertrigo, irritation cutanée, dermatite de stase, bulle hémorragique | Éruption d’origine médicamenteuse, dermatite exfoliative généralisée, érythème polymorphe, éruption avec exfoliation, pemphigoïde, dermatite allergique, dermatite de contact, intertrigo, irritation cutanée, dermatite de stase |
Fréquence indéterminée1 | Nécrolyse épidermique toxique, hyperpigmentation cutanée, décoloration cutanée, troubles de la pigmentation, syndrome de Stevens-Johnson, nécrose épidermique, exanthème intertrigineux et de flexion symétrique lié au médicament | Nécrolyse épidermique toxique, hyperpigmentation cutanée, décoloration cutanée, troubles de la pigmentation, syndrome de Stevens-Johnson, nécrose épidermique, exanthème intertrigineux et de flexion symétrique lié au médicament |
Affections musculo-squelettiques et systémiques | ||
Peu fréquent |
| Myosite |
Troubles généraux et anomalies au site d’administration | ||
Très fréquent | Fatigue | Fatigue |
Fréquent | Extravasation au site de perfusion | Extravasation au site de perfusion |
Investigations | ||
Très fréquent | Augmentation du taux d’alanine aminotransférase, augmentation du taux d’aspartate aminotransférase, perte de poids | Augmentation du taux d’alanine aminotransférase, augmentation du taux d’aspartate aminotransférase, perte de poids |
Fréquent |
| Augmentation du taux de lipase |
Lésions, intoxications et complication d’interventions | ||
Fréquent | Réactions liées à la perfusion | Réactions liées à la perfusion |
1Issus de l'expérience post-commercialisation mondiale.
2Inclut : syndrome de détresse respiratoire aiguë, maladie pulmonaire auto-immune, maladie pulmonaire à médiation immunitaire, pneumopathie interstitielle diffuse, opacité pulmonaire, pneumonie organisée, pneumopathie inflammatoire, fibrose pulmonaire, toxicité pulmonaire, sarcoïdose pulmonaire et sarcoïdose.
Description de certains effets indésirables
Immunogénicité
Un total de 159 patients a fait l’objet de test d’immunogénicité à l’enfortumab vedotin après l’administration d’enfortumab vedotin en association avec le pembrolizumab pour le traitement de la TVIM ; 3 patients ont été confirmés positifs à l'inclusion aux anticorps anti‑médicaments (AMA), et chez les patients qui étaient négatifs à l'inclusion (n = 156), 2 patients (1,3 %) étaient positifs après l’inclusion.
Un total de 490 patients a fait l’objet de test d’immunogénicité à l’enfortumab vedotin après l’administration d’enfortumab vedotin en association avec le pembrolizumab pour le traitement du carcinome urothélial non résécable ou métastatique ; 24 patients ont été confirmés positifs à l'inclusion aux AMA, et chez les patients qui étaient négatifs à l'inclusion (n = 466), 14 patients (3 %) étaient positifs après l'inclusion.
Un total de 697 patients a fait l’objet de test d’immunogénicité à l’enfortumab vedotin 1,25 mg/kg en monothérapie ; 16 patients ont été confirmés positifs à l'inclusion aux anticorps anti‑médicaments (AMA), et chez les patients qui étaient négatifs à l'inclusion (n = 681), 24 patients (3,5 %) étaient positifs après l'inclusion.
L’incidence de formation d’anticorps anti-enfortumab vedotin apparus sous traitement a été cohérente lorsqu'elle était évaluée après l’administration d’enfortumab vedotin en monothérapie et en association avec le pembrolizumab.
En raison du nombre limité de patients présentant des anticorps dirigés contre Padcev, aucune conclusion ne peut être tirée en ce qui concerne un effet potentiel de l'immunogénicité sur l'efficacité, la sécurité ou la pharmacocinétique.
Réactions cutanées
Dans les études cliniques portant sur l’enfortumab vedotin en association avec le pembrolizumab, des réactions cutanées sont survenues chez 89 % (648) des 731 patients, et, dans la majorité des cas, il s’agissait d’une éruption maculo-papuleuse, d’une éruption maculeuse, d’une éruption cutanée et d’une éruption papuleuse. Des réactions cutanées sévères (grade 3 ou 4) sont survenues chez 19 % (135) des patients (Grade 3 : 17%, Grade 4 : 2%). Un évènement d’issue fatale de nécrolyse épidermique toxique est survenu chez un patient. Le délai médian d’apparition des réactions cutanées sévères était de 1,6 mois (intervalle : 0,1 à 17,2 mois). Parmi les patients qui ont présenté des réactions cutanées et pour lesquels des données concernant la résolution de l’événement étaient disponibles (n = 496), la résolution complète des réactions cutanées a été rapportée chez 76 % des patients, une amélioration partielle a été rapportée chez 16 % des patients et aucune amélioration lors de la dernière évaluation n’a été rapportée chez 8 % des patients. Parmi les 24 % de patients qui présentaient des réactions cutanées résiduelles lors de la dernière évaluation, 25 % ont présenté des événements de grade ≥ 2.
Dans les études cliniques de l’enfortumab vedotin en monothérapie, des réactions cutanées sont survenues chez 57 % (452) des 793 patients traités par enfortumab vedotin 1,25 mg/kg. Des réactions cutanées sévères (grade 3 ou 4) sont survenues chez 14 % (108) des patients, et, dans la majorité des cas, il s’agissait d’éruption maculo-papuleuse, de stomatite, d’éruption érythémateuse, d’éruption d'origine médicamenteuse. Le délai médian d'apparition des réactions cutanées sévères était de 0,7 mois (intervalle : 0,1 à 8,2 mois). Des réactions cutanées graves sont survenues chez 4,3 % (34) des patients. Parmi les patients qui ont présenté des réactions cutanées et pour lesquels des données concernant la résolution de l’événement étaient disponibles (n = 366), la résolution complète des réactions cutanées a été rapportée chez 61 % des patients, une amélioration partielle a été rapportée chez 24 % des patients et aucune amélioration lors de la dernière évaluation n’a été rapportée chez 15 % des patients. Parmi les 39 % de patients qui présentaient des réactions cutanées résiduelles lors de la dernière évaluation, 38 % ont présenté des événements de grade ≥ 2.
Pneumopathie inflammatoire/PID
Dans les études cliniques portant sur l’enfortumab vedotin en association avec le pembrolizumab, une pneumopathie inflammatoire/PID est survenue chez 9,4% (69) des 731 patients. Une pneumopathie/PID sévère (grade 3 ou 4) est survenue chez 20 patients (grade 3 : 2,3 % et grade 4 : 0,4 %). La pneumopathie inflammatoire/PID a entraîné l’arrêt de l’enfortumab vedotin chez 2,1 % des patients. Trois patients ont présenté un événement de pneumopathie inflammatoire/PID d’issue fatale. Le délai médian de survenue d'une pneumopathie inflammatoire/PID, quel que soit le grade, était de 4,1 mois (intervalle : 0,3 à 26,2 mois).
Dans les études cliniques de l’enfortumab vedotin en monothérapie, une pneumopathie inflammatoire/PID est survenue chez 3,3% (26) des 793 patients traités par enfortumab vedotin 1,25 mg/kg. Moins de 1 % des patients ont présenté une pneumopathie inflammatoire sévère (grade 3 ou 4)/PID sévère (grade 3 : 0,5 %, grade 4 : 0,3 %). La pneumopathie inflammatoire/PID a conduit à l’arrêt du traitement par enfortumab vedotin chez 0,5 % des patients. Aucun décès n'est survenu à la suite d'une pneumopathie inflammatoire/PID. Le délai médian d'apparition d'une pneumopathie inflammatoire/PID, quel que soit le grade, était de 2,7 mois (intervalle : 0,6 à 6,0 mois) et la durée médiane de la pneumopathie inflammatoire/PID était de 1,6 mois (intervalle : 0,1 à 43,0 mois). Sur les 26 patients ayant présenté une pneumopathie inflammatoire/PID, les symptômes ont été résolus pour 8 (30,8 %) d’entre eux.
Hyperglycémie
Dans les études cliniques de l’enfortumab vedotin en monothérapie, une hyperglycémie (glycémie > 13,9 mmol/L) est survenue chez 17 % (133) des 793 patients traités par enfortumab vedotin 1,25 mg/kg. Des événements graves d’hyperglycémie sont survenus chez 2,5 % des patients, 7 % des patients ont développé une hyperglycémie sévère (grade 3 ou 4) et 0,3% des patients ont présenté des évènements d’issue fatale, parmi lesquels un cas d’hyperglycémie et un cas d’acidocétose diabétique. L'incidence de l'hyperglycémie de grade 3‑4 a augmenté de manière constante chez les patients avec un indice de masse corporelle plus élevé ainsi que chez les patients présentant une hémoglobine A1c plus élevée à l’inclusion (HbA1c). Le délai médian de survenue de l'hyperglycémie était de 0,5 mois (intervalle : 0 à 20,3 mois). Parmi les patients qui ont présenté une hyperglycémie et pour lesquels des données concernant la résolution de l’événement étaient disponibles (n = 106), la résolution de l’hyperglycémie a été rapportée chez 66 % des patients, une amélioration partielle a été rapportée chez 19 % des patients et aucune amélioration lors de la dernière évaluation n’a été rapportée chez 15 % des patients. Parmi les 34 % de patients ayant présenté une hyperglycémie résiduelle lors de la dernière évaluation, 64 % ont présenté des événements de grade ≥ 2.
Neuropathie périphérique
Dans les études cliniques de l’enfortumab vedotin en monothérapie, une neuropathie périphérique est survenue chez 53 % (422) des 793 patients traités par enfortumab vedotin 1,25 mg/kg. Cinq pour cent des patients ont présenté une neuropathie périphérique sévère (grade 3 ou 4), incluant des manifestations sensitives et motrices. Le délai médian de survenue d’une neuropathie périphérique de grade ≥ 2 était de 5 mois (intervalle : 0,1 à 20,2 mois). Parmi les patients qui ont présenté une neuropathie et pour lesquels des données concernant la résolution de l’événement étaient disponibles (n = 340), la résolution de la neuropathie a été rapportée chez 14 % des patients, une amélioration partielle a été rapportée chez 46 % des patients et aucune amélioration lors de la dernière évaluation n’a été rapportée chez 41 % des patients. Parmi les 86 % de patients qui avaient une neuropathie résiduelle lors de la dernière évaluation, 51 % ont présenté des événements de grade ≥ 2.
Affections oculaires
Dans les études cliniques de l’enfortumab vedotin en monothérapie, 30 % des patients ont présenté une sécheresse oculaire pendant le traitement par enfortumab vedotin 1,25 mg/kg. Le traitement a été interrompu chez 1,5 % des patients et 0,1 % des patients ont définitivement arrêté le traitement en raison d’une sécheresse oculaire. Une sécheresse oculaire sévère (grade 3) est survenue uniquement chez 3 patients (0,4 %). Le délai médian de survenue de la sécheresse oculaire était de 1,7 mois (intervalle : 0 à 30,6 mois).
Populations particulières
Personnes âgées
L’enfortumab vedotin, en association avec le pembrolizumab a été évalué chez 202 patients âgés de moins de 65 ans et chez 529 patients âgés de 65 ans et plus. En général, la fréquence des évènements indésirables était plus élevée chez les patients âgés de ≥ 65 ans que chez les patients âgés de < 65 ans, en particulier pour les évènements indésirables graves (58,8 % et 40,1 %, respectivement) et les évènements de grade ≥ 3 (79,8 % et 65,3 %, respectivement).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration.
Belgique
Agence fédérale des médicaments et des produits de santé
www.afmps.be
Division Vigilance:
Site internet: www.notifieruneffetindesirable.be
e-mail: adr@fagg-afmps.be
Luxembourg
Centre Régional de Pharmacovigilance de Nancy ou Division de la
pharmacie et des médicaments de la Direction de la santé
Site internet : www.guichet.lu/pharmacovigilance
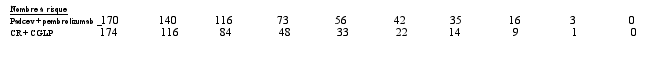
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Astellas Pharma Europe B.V.
Sylviusweg 62
2333 BE Leiden
Pays-Bas
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/21/1615/001
EU/1/21/1615/002
10. DATE DE MISE À JOUR DU TEXTE
19/06/2026
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 4566196 | PADCEV 20MG PDR SOL PERF FL 1 | - | € 600 | Oui | - | - | |
| 4566204 | PADCEV 30MG PDR SOL PERF FL 1 | - | € 900 | Oui | - | - |