1. DÉNOMINATION DU MÉDICAMENT
KEYTRUDA 25 mg/mL solution à diluer pour perfusion.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un flacon de 4 mL de solution à diluer contient 100 mg de pembrolizumab.
Chaque mL de solution à diluer contient 25 mg de pembrolizumab.
Pembrolizumab est un anticorps monoclonal humanisé (IgG4 isotype kappa avec altération stabilisatrice de séquence dans la région Fc) anti-PD-1 (programmed cell death-1), produit dans des cellules d’ovaires de hamster chinois par la technique de l’ADN recombinant.
Excipient à effet notoire :
Ce médicament contient 0,2 mg de polysorbate 80 dans chaque mL de solution à diluer.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Solution à diluer pour perfusion.
Solution limpide à légèrement opalescente, incolore à légèrement jaune, pH 5,2 – 5,8.
4. DONNÉES CLINIQUES
4.1 Indications thérapeutiques
Mélanome
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et des adolescents âgés de 12 ans et plus atteints d’un mélanome avancé (non résécable ou métastatique).
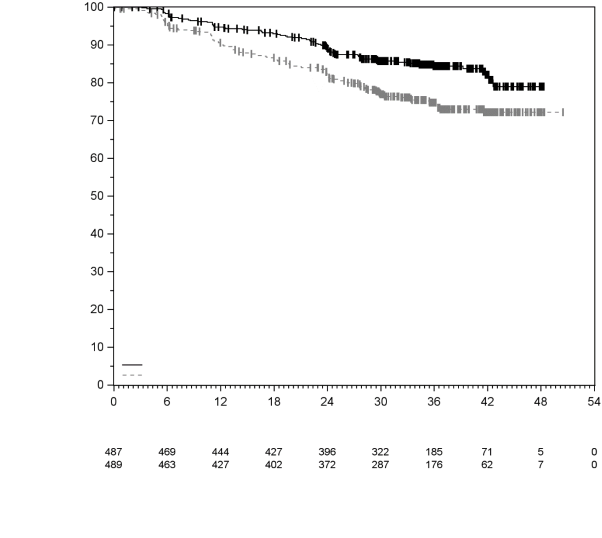
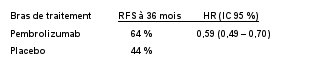
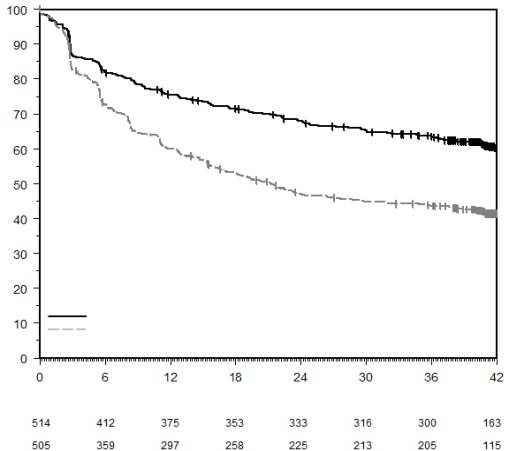
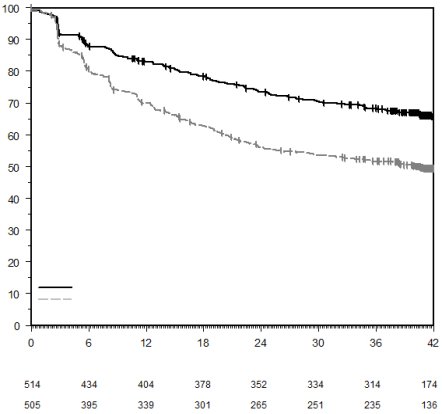
KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes et des adolescents âgés de 12 ans et plus atteints d’un mélanome de stade IIB, IIC ou III, ayant eu une résection complète (voir rubrique 5.1).
Cancer bronchique non à petites cellules (CBNPC)
KEYTRUDA, en association à une chimiothérapie à base de sels de platine en traitement néoadjuvant, puis poursuivi en monothérapie en traitement adjuvant, est indiqué dans le traitement des patients adultes atteints d’un cancer bronchique non à petites cellules résécable à haut risque de récidive (pour les critères de sélection, voir rubrique 5.1).
KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes atteints d’un cancer bronchique non à petites cellules à haut risque de récidive après résection complète et une chimiothérapie à base de sels de platine (pour les critères de sélection, voir rubrique 5.1).
KEYTRUDA est indiqué en monothérapie dans le traitement de première ligne des patients adultes atteints d’un cancer bronchique non à petites cellules métastatique dont les tumeurs expriment PD-L1 avec un score de proportion tumorale (TPS) 50 %, sans mutations tumorales d’EGFR ou d’ALK.
KEYTRUDA, en association à une chimiothérapie pemetrexed et sel de platine, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique non-épidermoïde dont les tumeurs ne présentent pas de mutations d’EGFR ou d’ALK.
KEYTRUDA, en association au carboplatine et au paclitaxel ou au nab-paclitaxel, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique épidermoïde.
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints de cancer bronchique non à petites cellules localement avancé ou métastatique dont les tumeurs expriment PD-L1 avec un TPS 1 %, et ayant reçu au moins une chimiothérapie antérieure. Les patients présentant des mutations tumorales d’EGFR ou d’ALK doivent également avoir reçu une thérapie ciblée avant de recevoir KEYTRUDA.
Mésothéliome pleural malin (MPM)
KEYTRUDA, en association à une chimiothérapie pemetrexed et sel de platine, est indiqué dans le traitement de première ligne des patients adultes atteints d’un mésothéliome pleural malin non épithélioïde non résécable.
Lymphome de Hodgkin classique (LHc)
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et pédiatriques âgés de 3 ans et plus atteints d’un lymphome de Hodgkin classique en rechute ou réfractaire après échec d’une greffe de cellules souches (GCS) autologue ou après au moins deux lignes de traitement antérieures lorsque la GCS autologue n’est pas une option de traitement.
Carcinome urothélial
KEYTRUDA, en association à l'enfortumab vedotin, est indiqué dans le traitement de première ligne des patients adultes atteints d’un carcinome urothélial non résécable ou métastatique.
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome urothélial localement avancé ou métastatique ayant reçu une chimiothérapie antérieure à base de sels de platine (voir rubrique 5.1).
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome urothélial localement avancé ou métastatique inéligibles à une chimiothérapie à base de cisplatine et dont les tumeurs expriment PD-L1 avec un score positif combiné (CPS) ≥ 10 (voir rubrique 5.1).
Carcinome épidermoïde de la tête et du cou (CETEC)
KEYTRUDA est indiqué en monothérapie en traitement néoadjuvant, poursuivi en traitement adjuvant en association à une radiothérapie avec ou sans cisplatine concomitant puis en monothérapie chez les patients adultes atteints d’un carcinome épidermoïde de la tête et du cou localement avancé résécable dont les tumeurs expriment PD-L1 avec un CPS ≥ 1.
KEYTRUDA est indiqué en monothérapie ou en association à une chimiothérapie à base de sels de platine et de 5-fluorouracile (5-FU) dans le traitement de première ligne des patients adultes atteints d’un carcinome épidermoïde de la tête et du cou métastatique ou récidivant non résécable dont les tumeurs expriment PD-L1 avec un CPS ≥ 1 (voir rubrique 5.1).
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d’un carcinome épidermoïde de la tête et du cou récidivant ou métastatique dont les tumeurs expriment PD-L1 avec un TPS ≥ 50 % et en progression pendant ou après une chimiothérapie à base de sels de platine (voir rubrique 5.1).
Carcinome à cellules rénales (CCR)
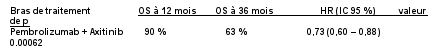
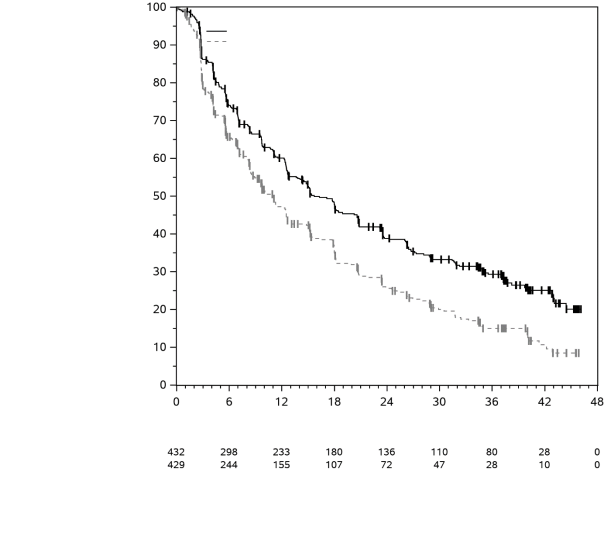
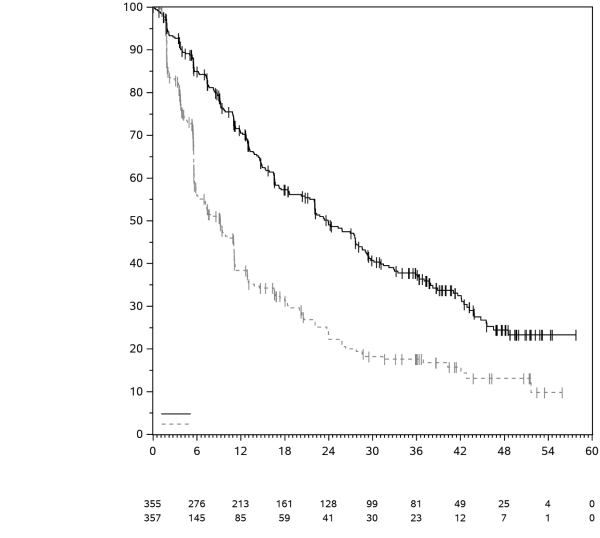
KEYTRUDA, en association à l’axitinib, est indiqué dans le traitement de première ligne des patients adultes atteints d’un carcinome à cellules rénales avancé (voir rubrique 5.1).
KEYTRUDA, en association au lenvatinib, est indiqué dans le traitement de première ligne des patients adultes atteints d’un carcinome à cellules rénales avancé (voir rubrique 5.1).
KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes atteints d’un carcinome à cellules rénales à risque accru de récidive post néphrectomie, ou après une néphrectomie et une résection des lésions métastatiques (pour les critères de sélection, voir rubrique 5.1).
Cancers avec instabilité microsatellitaire élevée (MSI-H) ou déficience du système de réparation des mésappariements de l’ADN (dMMR)
Cancer colorectal
KEYTRUDA est indiqué en monothérapie chez des patients adultes atteints d’un cancer colorectal MSI-H ou dMMR aux stades suivants :
- traitement de première ligne d’un cancer colorectal métastatique ;
- traitement d’un cancer colorectal non résécable ou métastatique après traitement antérieur à base de fluoropyrimidine en association.
Cancers non-colorectaux
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints de tumeurs MSI-H ou dMMR suivantes :
- cancer de l'endomètre avancé ou récidivant, dont la maladie progresse pendant ou après un traitement antérieur à base de sels de platine reçu quel que soit le stade et qui ne sont pas éligibles à une chirurgie curative ou à une radiothérapie ;
- cancer gastrique, de l'intestin grêle ou des voies biliaires non résécable ou métastatique, dont la maladie progresse pendant ou après au moins un traitement antérieur.
Cancer de l'œsophage
KEYTRUDA, en association à une chimiothérapie à base de sels de platine et de fluoropyrimidine, est indiqué dans le traitement de première ligne des patients adultes atteints d'un cancer de l'œsophage localement avancé non résécable ou métastatique, dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 ( voir section 5.1).
Cancer du sein triple négatif (CSTN)
KEYTRUDA, en association à une chimiothérapie comme traitement néoadjuvant, puis poursuivi après la chirurgie en monothérapie comme traitement adjuvant, est indiqué dans le traitement des patients adultes atteints d'un cancer du sein triple négatif localement avancé ou de stade précoce à haut risque de récidive (voir rubrique 5.1).
KEYTRUDA, en association à une chimiothérapie, est indiqué dans le traitement des patients adultes atteints d’un cancer du sein triple négatif localement récurrent non résécable ou métastatique, dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 et qui n'ont pas reçu de chimiothérapie antérieure pour la maladie métastatique (voir section 5.1).
Cancer de l’endomètre (CE)
KEYTRUDA, en association au carboplatine et au paclitaxel, est indiqué dans le traitement de première ligne des patientes adultes atteintes d’un cancer de l’endomètre avancé nouvellement diagnostiqué ou récidivant qui sont éligibles à un traitement systémique.
KEYTRUDA, en association au lenvatinib, est indiqué dans le traitement des patientes adultes atteintes d’un cancer de l’endomètre avancé ou récidivant, dont la maladie progresse pendant ou après un traitement antérieur à base de sels de platine reçu quel que soit le stade et qui ne sont pas éligibles à une chirurgie curative ou à une radiothérapie.
Cancer du col de l'utérus
KEYTRUDA, en association à la radiochimiothérapie (radiothérapie externe suivie d'une curiethérapie), est indiqué dans le traitement des patientes adultes atteintes d’un cancer du col de l'utérus localement avancé de Stade III - IVA selon FIGO 2014, qui n’ont pas reçu de traitement définitif préalable.
KEYTRUDA, en association à une chimiothérapie avec ou sans bevacizumab, est indiqué dans le traitement des patientes adultes atteintes d’un cancer du col de l'utérus persistant, récidivant ou métastatique, dont les tumeurs expriment PD-L1 avec un CPS ≥ 1.
Adénocarcinome gastrique ou de la jonction œso-gastrique (JOG)
KEYTRUDA, en association au trastuzumab et à une chimiothérapie à base de sels de platine et de fluoropyrimidine, est indiqué dans le traitement de première ligne des patients adultes atteints d’un adénocarcinome gastrique ou de la jonction œso-gastrique, localement avancé non résécable ou métastatique, HER-2 positif et dont les tumeurs expriment PD-L1 avec un CPS ≥ 1.
KEYTRUDA, en association à une chimiothérapie à base de sels de platine et de fluoropyrimidine, est indiqué dans le traitement de première ligne des patients adultes atteints d’un adénocarcinome gastrique ou de la jonction œso-gastrique, localement avancé non résécable ou métastatique, HER-2 négatif et dont les tumeurs expriment PD-L1 avec un CPS ≥ 1 (voir section 5.1).
Carcinome des voies biliaires (CVB)
KEYTRUDA, en association à la gemcitabine et au cisplatine, est indiqué dans le traitement de première ligne des patients adultes atteints d’un carcinome des voies biliaires localement avancé non résécable ou métastatique.
4.2 Posologie et mode d’administration
Le traitement doit être initié et supervisé par des médecins qualifiés et expérimentés dans l’utilisation de traitements anticancéreux.
Les patients recevant pembrolizumab sous-cutané peuvent passer au pembrolizumab intraveineux lors de leur prochaine dose programmée. Les patients recevant pembrolizumab intraveineux peuvent passer au pembrolizumab sous-cutané lors de leur prochaine dose programmée.
Test PD-L1
Si cela est spécifié dans l'indication, la sélection des patients pour le traitement par KEYTRUDA basée sur l'expression tumorale de PD-L1 doit être confirmée par un test validé (voir rubriques 4.1, 4.4, 4.8 et 5.1).
Test MSI/MMR
Si cela est spécifié dans l'indication, la sélection des patients pour le traitement par KEYTRUDA basée sur le statut tumoral MSI-H/dMMR doit être confirmée par un test validé (voir rubriques 4.1 et 5.1).
Posologie
La dose recommandée de KEYTRUDA chez les adultes est soit de 200 mg toutes les 3 semaines, soit de 400 mg toutes les 6 semaines, administrée en perfusion intraveineuse pendant 30 minutes.
La dose recommandée de KEYTRUDA en monothérapie chez les patients pédiatriques âgés de 3 ans et plus atteints d’un LHc ou chez les patients âgés de 12 ans et plus atteints d’un mélanome est de 2 mg/kg de poids corporel (jusqu’à un maximum de 200 mg) toutes les 3 semaines, administrée en perfusion intraveineuse pendant 30 minutes.
Pour une utilisation en association, voir le Résumé des Caractéristiques du Produit (RCP) des traitements concomitants.
Les patients doivent être traités par KEYTRUDA jusqu’à progression de la maladie ou toxicité inacceptable (et jusqu'à la durée maximale du traitement si spécifiée pour une indication). Des réponses atypiques (c’est-à-dire une augmentation initiale et transitoire de la taille de la tumeur ou l’apparition de nouvelles lésions de petite taille durant les premiers mois, suivies d’une régression de la tumeur) ont été observées. Chez les patients cliniquement stables présentant une progression initiale de la maladie, il est recommandé de poursuivre le traitement jusqu'à ce que la progression soit confirmée.
Dans le traitement adjuvant du mélanome, du CBNPC ou du carcinome à cellules rénales, KEYTRUDA doit être administré jusqu’à récidive de la maladie, toxicité inacceptable ou pendant une durée allant jusqu’à un an.
Dans le traitement néoadjuvant et adjuvant du CBNPC résécable, les patients doivent être traités par KEYTRUDA en néoadjuvant en association à une chimiothérapie à raison de 4 doses de 200 mg toutes les 3 semaines ou 2 doses de 400 mg toutes les 6 semaines ou jusqu'à progression de la maladie empêchant une chirurgie définitive ou toxicité inacceptable, suivi d'un traitement adjuvant par KEYTRUDA en monothérapie à raison de 13 doses de 200 mg toutes les 3 semaines ou 7 doses de 400 mg toutes les 6 semaines ou jusqu'à récidive de la maladie ou toxicité inacceptable. Les patients dont la progression de la maladie empêche une chirurgie définitive ou qui présentent une toxicité inacceptable liée à KEYTRUDA en traitement néoadjuvant en association à une chimiothérapie ne doivent pas recevoir KEYTRUDA en monothérapie en traitement adjuvant.
Dans le traitement néoadjuvant et adjuvant du CETEC localement avancé résécable, les patients doivent être traités par KEYTRUDA en néoadjuvant en monothérapie à raison de 2 doses de 200 mg toutes les 3 semaines ou 1 dose de 400 mg ou jusqu’à progression de la maladie empêchant une chirurgie définitive ou toxicité inacceptable, suivi d’un traitement adjuvant par KEYTRUDA en association à une radiothérapie avec ou sans cisplatine concomitant à raison de 3 doses de 200 mg toutes les 3 semaines ou 2 doses de 400 mg toutes les 6 semaines, suivi par KEYTRUDA en monothérapie à raison de 12 doses de 200 mg toutes les 3 semaines ou 6 doses de 400 mg toutes les 6 semaines ou jusqu’à récidive de la maladie ou toxicité inacceptable. Les patients dont la progression de la maladie empêche une chirurgie définitive ou qui présentent une toxicité inacceptable liée à KEYTRUDA en traitement néoadjuvant en monothérapie ne doivent pas recevoir KEYTRUDA en association à une radiothérapie avec ou sans cisplatine concomitant en traitement adjuvant.
Dans le traitement néoadjuvant et adjuvant du CSTN, les patients doivent être traités par KEYTRUDA en néoadjuvant en association à une chimiothérapie à raison de 8 doses de 200 mg toutes les 3 semaines ou 4 doses de 400 mg toutes les 6 semaines ou jusqu'à progression de la maladie empêchant une chirurgie définitive ou toxicité inacceptable, suivi d'un traitement adjuvant par KEYTRUDA en monothérapie à raison de 9 doses de 200 mg toutes les 3 semaines ou 5 doses de 400 mg toutes les 6 semaines ou jusqu'à récidive de la maladie ou toxicité inacceptable. Les patients dont la progression de la maladie empêche une chirurgie définitive ou qui présentent une toxicité inacceptable liée à KEYTRUDA en traitement néoadjuvant en association à une chimiothérapie ne doivent pas recevoir KEYTRUDA en monothérapie en traitement adjuvant.
Dans le cancer du col de l'utérus localement avancé, les patientes doivent être traitées par KEYTRUDA en concomitance à une radiochimiothérapie, suivi de KEYTRUDA en monothérapie. KEYTRUDA peut être administré à raison de 200 mg toutes les 3 semaines ou de 400 mg toutes les 6 semaines jusqu'à progression de la maladie, toxicité inacceptable ou jusqu'à 24 mois.
Suspension ou arrêt définitif du traitement (voir aussi rubrique 4.4)
Aucune réduction de dose de KEYTRUDA n’est recommandée. KEYTRUDA doit être suspendu ou arrêté pour gérer les effets indésirables tels que décrit dans le tableau 1.
Tableau 1 : Modifications de traitement recommandées pour KEYTRUDA
Effets indésirables à médiation immunitaire | Sévérité | Modification de traitement |
Pneumopathie inflammatoire | Grade 2 | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Grades 3 ou 4, ou Grade 2 récurrent | Arrêt définitif | |
Colite | Grades 2 ou 3 | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Grade 4 ou Grade 3 récurrent | Arrêt définitif | |
Néphrite | Grade 2 avec créatinine > 1,5 à ≤ 3 fois la limite supérieure de la normale (LSN) | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Grade ≥ 3 avec créatinine > 3 fois la LSN | Arrêt définitif | |
Endocrinopathies | Insuffisance surrénalienne et hypophysite de Grade 2 | Suspension du traitement jusqu’au contrôle par traitement hormonal substitutif |
Insuffisance surrénalienne ou hypophysite symptomatique de Grades 3 ou 4 | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* | |
Hypothyroïdie | L’hypothyroïdie peut être prise en charge par traitement hormonal substitutif sans interruption du traitement. | |
Hépatite | Grade 2 avec aspartate aminotransférase (ASAT) ou alanine aminotransférase (ALAT) > 3 à 5 fois la LSN ou bilirubine totale > 1,5 à 3 fois la LSN | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Grade ≥ 3 avec ASAT ou ALAT > 5 fois la LSN ou bilirubine totale > 3 fois la LSN | Arrêt définitif | |
En cas de métastases hépatiques avec une augmentation initiale de Grade 2 des ASAT ou des ALAT, hépatite avec augmentation des ASAT ou des ALAT ≥ 50 % pendant ≥ 1 semaine | Arrêt définitif | |
Réactions cutanées | Grade 3 ou syndrome de Stevens-Johnson (SSJ) ou nécrolyse épidermique toxique (NET) suspectés | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Grade 4 ou SSJ ou NET confirmés | Arrêt définitif | |
Autres effets indésirables à médiation immunitaire | Selon la sévérité et le type de réaction (Grade 2 ou Grade 3) | Suspension jusqu’à amélioration des effets indésirables aux Grades 0-1* |
Myocardite de Grades 3 ou 4 | Arrêt définitif | |
Grade 4 ou Grade 3 récurrent | Arrêt définitif | |
Réactions liées à la perfusion | Grades 3 ou 4 | Arrêt définitif |
Note : Les grades de toxicité sont en accord avec la terminologie de l’US National Cancer Institute – Common Terminology Criteria for Adverse Event version 4.0 (NCI-CTCAE v.4)
* Si une toxicité liée au traitement ne s’améliore pas jusqu’aux Grades 0-1 dans les 12 semaines après la dernière administration de KEYTRUDA, ou si la dose de corticostéroïdes ne peut pas être réduite dans les 12 semaines à une dose ≤ 10 mg de prednisone ou équivalent par jour, KEYTRUDA doit être arrêté définitivement.
La sécurité de la ré-administration d’un traitement par pembrolizumab chez les patients ayant précédemment présenté une myocardite à médiation immunitaire n’est pas connue.
KEYTRUDA, en monothérapie ou en association, doit être arrêté définitivement en cas d’effets indésirables à médiation immunitaire de Grade 4 ou de Grade 3 récurrent, sauf indication contraire dans le Tableau 1.
En cas de toxicité hématologique de Grade 4, uniquement chez les patients atteints d’un LHc, KEYTRUDA doit être suspendu jusqu’à amélioration des effets indésirables aux Grades 0-1.
KEYTRUDA en association à l’axitinib dans le CCR
Chez les patients atteints d’un CCR traités par KEYTRUDA en association à l’axitinib, voir le RCP concernant la posologie de l’axitinib. En association à pembrolizumab, l’augmentation de dose d’axitinib au-delà de la dose initiale de 5 mg peut être envisagée à intervalles de six semaines ou plus (voir rubrique 5.1).
En cas d’augmentation des enzymes hépatiques chez les patients atteints d’un CCR traités par KEYTRUDA en association à l’axitinib :
- Si les ALAT ou les ASAT sont ≥ 3 fois la LSN mais < 10 fois la LSN sans bilirubine totale concomitante ≥ 2 fois la LSN, KEYTRUDA et l’axitinib doivent être suspendus jusqu’à amélioration de ces effets indésirables jusqu’aux Grades 0-1. Une corticothérapie peut être envisagée. La réintroduction d’un seul médicament ou la réintroduction séquentielle des deux médicaments après amélioration peut être envisagée. En cas de réintroduction de l’axitinib, une réduction de la dose peut être envisagée comme mentionné dans le RCP de l’axitinib.
- Si les ALAT ou les ASAT sont ≥ 10 fois la LSN ou > 3 fois la LSN avec une bilirubine totale concomitante ≥ 2 fois la LSN, KEYTRUDA et l’axitinib doivent être arrêtés définitivement et une corticothérapie peut être envisagée.
KEYTRUDA en association au lenvatinib
Lorsqu'il est utilisé en association au lenvatinib, l’un ou les deux médicaments doivent être interrompus selon le cas. Lenvatinib doit être suspendu, sa dose doit être réduite ou il doit être arrêté conformément aux instructions concernant l’association au pembrolizumab dans le RCP du lenvatinib . Aucune réduction de dose n'est recommandée pour KEYTRUDA.
Les patients traités par KEYTRUDA doivent avoir reçu la carte patient et avoir été informés des risques de KEYTRUDA (voir également la notice).
Populations particulières
Personnes âgées
Aucune adaptation posologique n’est nécessaire chez les patients âgés de ≥ 65 ans (voir rubriques 4.4 et 5.1).
Insuffisance rénale
Aucune adaptation posologique n’est nécessaire pour les patients présentant une insuffisance rénale légère ou modérée. KEYTRUDA n’a pas été étudié chez les patients présentant une insuffisance rénale sévère (voir rubriques 4.4 et 5.2).
Insuffisance hépatique
Aucune adaptation posologique n’est nécessaire pour les patients présentant une insuffisance hépatique légère ou modérée. KEYTRUDA n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité de KEYTRUDA chez les enfants de moins de 18 ans n’ont pas été établies, sauf pour les patients pédiatriques atteints d’un mélanome ou d’un LHc. Les données actuellement disponibles sont décrites en rubriques 4.8, 5.1 et 5.2.
Mode d’administration
Il est important de vérifier l’étiquette du flacon pour s’assurer que la formulation correcte (intraveineuse ou sous-cutanée) est préparée et administrée au patient comme prescrit, pour réduire le risque d’erreurs médicamenteuses.
KEYTRUDA solution à diluer pour perfusion est à usage intraveineux uniquement. KEYTRUDA solution à diluer pour perfusion n’est pas destiné à une administration sous-cutanée.
KEYTRUDA solution à diluer pour perfusion doit être administré par perfusion sur une durée de 30 minutes. La formulation intraveineuse de KEYTRUDA ne doit pas être administrée en injection rapide ou en bolus.
KEYTRUDA solution à diluer pour perfusion ne doit pas être substitué à ou remplacé par pembrolizumab sous-cutané, car leurs dosages et voies d’administration recommandés diffèrent.
Lorsque KEYTRUDA est utilisé en association à une chimiothérapie intraveineuse, KEYTRUDA doit être administré en premier.
Lorsque KEYTRUDA est utilisé en association à l'enfortumab vedotin, KEYTRUDA doit être administré après l'enfortumab vedotin lorsqu'il est administré le même jour.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique 6.6.
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité
Pembrolizumab est le plus fréquemment associé à des effets indésirables à médiation immunitaire. La plupart d’entre eux, y compris les réactions sévères, se sont résolus après initiation d’un traitement médical approprié ou arrêt de pembrolizumab (voir « Description d’une sélection d’effets indésirables » ci-dessous). Les fréquences mentionnées ci-dessous et dans le tableau 2 sont basées sur tous les effets indésirables rapportés, quelle que soit l’évaluation de la causalité par l’investigateur.
Pembrolizumab en monothérapie (voir rubrique 4.2)
La sécurité de pembrolizumab en monothérapie a été évaluée dans des études cliniques chez 7 631 patients dans différents types de tumeurs et avec quatre doses (2 mg/kg de poids corporel toutes les 3 semaines, 200 mg toutes les 3 semaines ou 10 mg/kg de poids corporel toutes les 2 ou 3 semaines). Dans cette population de patients, la durée d’observation médiane était de 8,5 mois (de 1 jour à 39 mois) et les effets indésirables les plus fréquents avec pembrolizumab étaient : fatigue (31 %), diarrhée (22 %) et nausée (20 %). La majorité des effets indésirables rapportés en monothérapie étaient d’une sévérité de Grades 1 ou 2. Les effets indésirables les plus graves étaient des effets indésirables à médiation immunitaire et des réactions sévères liées à la perfusion (voir rubrique 4.4). Les incidences des effets indésirables à médiation immunitaire étaient de 37 % tous Grades, et 9 % pour les Grades 3-5 avec le pembrolizumab en monothérapie au stade adjuvant, et 25 % tous Grades, et 6 % pour les Grades 3-5 au stade métastatique. Aucun nouvel effet indésirable à médiation immunitaire n'a été identifié au stade adjuvant.
Pembrolizumab en association à une chimiothérapie, à une radiothérapie (RT) ou à une radiochimiothérapie (RCT) (voir rubrique 4.2)
Lorsque pembrolizumab est administré en association, reportez-vous au RCP des médicaments respectifs du traitement en association avant l’initiation du traitement.
La sécurité de pembrolizumab en association à une chimiothérapie, une RT ou une RCT a été évaluée dans des études cliniques chez 6 695 patients dans différents types de tumeurs recevant 200 mg, 2 mg/kg de poids corporel ou 10 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines. Dans cette population de patients, les effets indésirables les plus fréquents étaient : nausées (51 %), anémie (50 %), diarrhées (35 %), fatigue (35 %), constipation (32 %), vomissements (27 %), diminution du nombre de neutrophiles (26 %) et diminution de l’appétit (26 %). Les incidences des effets indésirables de Grades 3-5 chez les patients avec un CBNPC étaient de 69 % pour le traitement par pembrolizumab en association et de 61 % pour la chimiothérapie seule, chez les patients avec un CETEC étaient de 80 % pour le traitement par pembrolizumab en association (chimiothérapie ou RT avec ou sans chimiothérapie) et de 79 % pour la chimiothérapie avec cétuximab ou RT avec ou sans chimiothérapie, chez les patients atteints d'un cancer de l'œsophage étaient de 86 % pour le traitement par pembrolizumab en association et de 83 % pour la chimiothérapie seule, chez les patients atteints d’un CSTN étaient de 80 % pour le traitement par pembrolizumab en association et de 77 % pour la chimiothérapie seule, chez les patientes atteintes d'un cancer du col de l'utérus étaient de 77 % pour le traitement par pembrolizumab en association (chimiothérapie avec ou sans bevacizumab ou en association à une RCT) et de 71 % pour la chimiothérapie avec ou sans bevacizumab ou la RCT seule, chez les patients atteints d'un cancer gastrique étaient de 74 % pour le traitement par pembrolizumab en association (chimiothérapie avec ou sans trastuzumab) et de 68 % pour la chimiothérapie avec ou sans trastuzumab, chez les patients atteints d’un carcinome des voies biliaires étaient de 85 % pour le traitement par pembrolizumab en association et 84 % pour la chimiothérapie seule, chez les patientes atteintes d’un CE étaient de 59 % pour le traitement par pembrolizumab en association et 46 % pour la chimiothérapie seule et chez les patients atteints d’un mésothéliome pleural malin étaient de 44 % pour le traitement par pembrolizumab en association et 30 % pour la chimiothérapie seule.
Pembrolizumab en association à un inhibiteur de tyrosine kinase (ITK) (voir rubrique 4.2)
Lorsque pembrolizumab est administré en association à l'axitinib ou au lenvatinib, reportez-vous au RCP de l'axitinib ou du lenvatinib avant l’initiation du traitement. Pour des informations complémentaires sur la sécurité du lenvatinib pour le carcinome à cellules rénales (CCR) avancé, voir le RCP de Kisplyx et pour le cancer de l’endomètre (CE) avancé, voir le RCP de Lenvima. Pour des informations complémentaires sur la sécurité de l'axitinib en cas d'élévation des enzymes hépatiques, voir également la rubrique 4.4.
La sécurité de pembrolizumab en association à l'axitinib ou au lenvatinib dans le CCR avancé, et en association au lenvatinib dans le CE avancé, a été évaluée chez un total de 1 456 patients atteints d’un CCR avancé ou d’un CE avancé recevant 200 mg de pembrolizumab toutes les 3 semaines avec soit 5 mg d’axitinib deux fois par jour, soit 20 mg de lenvatinib une fois par jour dans les études cliniques, selon le cas. Dans ces populations de patients, les effets indésirables les plus fréquents étaient : diarrhée (58 %), hypertension (54 %), hypothyroïdie (46 %), fatigue (41 %), diminution de l'appétit (40 %), nausées (40 %), arthralgie (30 %), vomissements (28 %), perte de poids (28 %), dysphonie (28 %), douleurs abdominales (28 %), protéinurie (27 %), syndrome main-pied (26 %), éruption cutanée (26 %), stomatite (25 %), constipation (25 %), douleurs musculosquelettiques (23 %), céphalées (23 %) et toux (21 %). La fréquence des effets indésirables de Grade 3-5 chez les patients atteints de CCR était de 80 % pour le pembrolizumab en association à l'axitinib ou au lenvatinib et de 71 % pour le sunitinib seul. Chez les patientes atteintes de CE, la fréquence des effets indésirables de Grade 3-5 était de 89 % pour le pembrolizumab en association au lenvatinib et de 73 % pour la chimiothérapie seule.
Résumé tabulé des effets indésirables
Les effets indésirables observés dans les études cliniques avec pembrolizumab en monothérapie ou en association avec la chimiothérapie, la RT ou la RCT ou d’autres médicaments anti-cancéreux, ou rapportés depuis la commercialisation de pembrolizumab sont listés dans le tableau 2. Ces effets sont présentés par classes de systèmes d’organes et par fréquence. Les fréquences sont définies ainsi : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Pour chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. Les effets indésirables connus pour survenir avec pembrolizumab ou les médicaments du traitement en association administrés seuls peuvent apparaître pendant le traitement avec ces médicaments en association, même si ces effets n'ont pas été rapportés au cours des études cliniques avec l’association thérapeutique.
Pour plus d'informations sur la sécurité lorsque pembrolizumab est administré en association, reportez-vous au RCP des médicaments respectifs du traitement en association.
Tableau 2 : Effets indésirables chez les patients traités par pembrolizumab†
Classification MedDRA par organe et par fréquence | Monothérapie | En association avec une chimiothérapie, une radiothérapie ou une radiochimiothérapie | En association avec axitinib ou lenvatinib |
Infections et infestations | |||
Très fréquent |
|
| infections des voies urinaires |
Fréquent | pneumonie | pneumonie | pneumonie |
Affections hématologiques et du système lymphatique | |||
Très fréquent | anémie | anémie, neutropénie, thrombopénie | anémie |
Fréquent | thrombopénie, neutropénie, lymphopénie | neutropénie fébrile, leucopénie, lymphopénie | neutropénie, thrombopénie, lymphopénie, leucopénie |
Peu fréquent | leucopénie, thrombopénie immunitaire, éosinophilie | anémie hémolytique⁎, éosinophilie | éosinophilie |
Rare | anémie hémolytique⁎, lymphohistiocytose hémophagocytaire, érythroblastopénie | thrombopénie immunitaire |
|
Affections du système immunitaire | |||
Fréquent | réaction liée à la perfusion⁎ | réaction liée à la perfusion⁎ | réaction liée à la perfusion⁎ |
Peu fréquent | sarcoïdose⁎ |
|
|
Rare |
| sarcoïdose |
|
Fréquence indéterminée | rejet de greffe d’organe solide |
|
|
Affections endocriniennes | |||
Très fréquent | hypothyroïdie⁎ | hypothyroïdie⁎ | hypothyroïdie |
Fréquent | hyperthyroïdie | insuffisance surrénalienne⁎, hyperthyroïdie⁎, thyroïdite⁎ | insuffisance surrénalienne⁎, hyperthyroïdie, thyroïdite⁎ |
Peu fréquent | insuffisance surrénalienne⁎, hypophysite⁎, | hypophysite⁎ | hypophysite⁎ |
Rare | hypoparathyroïdie | hypoparathyroïdie | hypoparathyroïdie |
Troubles du métabolisme et de la nutrition | |||
Très fréquent | diminution de l’appétit | hypokaliémie, diminution de l’appétit | diminution de l’appétit |
Fréquent | hyponatrémie, hypokaliémie, hypocalcémie | hyponatrémie, hypocalcémie | hyponatrémie, hypokaliémie, hypocalcémie |
Peu fréquent | diabète de type I⁎ | diabète de type I⁎ | diabète de type I⁎ |
Affections psychiatriques | |||
Très fréquent |
| insomnie |
|
Fréquent | insomnie |
| insomnie |
Affections du système nerveux | |||
Très fréquent | céphalée | neuropathie périphérique, céphalées | céphalée, dysgueusie |
Fréquent | étourdissements, neuropathie périphérique, léthargie, dysgueusie | étourdissements, dysgueusie | étourdissements, neuropathie périphérique, léthargie |
Peu fréquent | syndrome myasthénique⁎, épilepsie | encéphalite⁎, épilepsie, léthargie | syndrome myasthénique⁎, encéphalite⁎ |
Rare | syndrome de Guillain-Barré⁎, encéphalite⁎, myélite⁎, névrite optique, méningite (aseptique)⁎ | syndrome myasthénique⁎, syndrome de Guillain‑Barré⁎, myélite, névrite optique, méningite (aseptique) | névrite optique |
Affections oculaires | |||
Fréquent | sécheresse oculaire | sécheresse oculaire | sécheresse oculaire |
Peu fréquent | uvéite⁎ | uvéite⁎ | uvéite⁎ |
Rare | syndrome de Vogt-Koyanagi-Harada |
| syndrome de Vogt-Koyanagi-Harada |
Affections cardiaques | |||
Fréquent | arythmie cardiaque‡ (y compris fibrillation auriculaire) | arythmie cardiaque‡ (y compris fibrillation auriculaire) | arythmie cardiaque‡ (y compris fibrillation auriculaire) |
Peu fréquent | myocardite, péricardite⁎, épanchement péricardique | myocardite⁎, péricardite⁎, épanchement péricardique | myocardite, épanchement péricardique |
Affections vasculaires | |||
Très fréquent |
|
| hypertension |
Fréquent | hypertension | hypertension |
|
Peu fréquent |
| vascularite⁎ | vascularite⁎ |
Rare | vascularite⁎ |
|
|
Affections respiratoires, thoraciques et médiastinales | |||
Très fréquent | dyspnée, toux | dyspnée, toux | dyspnée, toux |
Fréquent | pneumopathie inflammatoire⁎ | pneumopathie inflammatoire⁎ | pneumopathie inflammatoire⁎ |
Affections gastro-intestinales | |||
Très fréquent | diarrhée, douleurs abdominales⁎, nausées, vomissements, constipation | diarrhée, nausées, vomissements, douleurs abdominales⁎, constipation | diarrhée, douleurs abdominales⁎, nausées, vomissements, constipation |
Fréquent | colite⁎, sécheresse buccale | colite⁎, gastrite⁎, sécheresse buccale | colite⁎, pancréatite⁎, gastrite⁎, sécheresse buccale |
Peu fréquent | pancréatite⁎, gastrite⁎, ulcération gastro-intestinale⁎ | pancréatite⁎, ulcération gastro-intestinale⁎ | ulcération gastro-intestinale⁎ |
Rare | insuffisance pancréatique exocrine, perforation de l’intestin grêle, maladie coeliaque | insuffisance pancréatique exocrine, perforation de l’intestin grêle, maladie coeliaque | perforation de l’intestin grêle |
Fréquence indéterminée |
|
| insuffisance pancréatique exocrine, maladie coeliaque |
Troubles hépatobiliaires | |||
Fréquent | hépatite⁎ | hépatite⁎ | hépatite⁎ |
Rare | cholangite sclérosante | cholangite sclérosante⁎ |
|
Affections de la peau et du tissu sous-cutané | |||
Très fréquent | prurit⁎, éruption cutanée⁎ | éruption cutanée⁎, alopécie, prurit⁎ | éruption cutanée⁎, prurit⁎ |
Fréquent | réactions cutanées sévères⁎, érythème, dermatite, sécheresse cutanée, vitiligo⁎, eczéma, alopécie, dermatite acnéiforme | réactions cutanées sévères⁎, érythème, dermatite, sécheresse cutanée, dermatite acnéiforme, eczéma | réactions cutanées sévères⁎, dermatite, sécheresse cutanée, érythème, dermatite acnéiforme, alopécie |
Peu fréquent | psoriasis, kératose lichénoïde⁎, papule, modification de la couleur des cheveux | psoriasis, kératose lichénoïde⁎, vitiligo⁎, papule | eczéma, kératose lichénoïde⁎, psoriasis, vitiligo⁎, papule, modification de la couleur des cheveux |
Rare | syndrome de Stevens-Johnson, érythème noueux, nécrolyse épidermique toxique | syndrome de Stevens-Johnson, érythème noueux, modification de la couleur des cheveux | nécrolyse épidermique toxique, syndrome de Stevens-Johnson |
Affections musculo-squelettiques et systémiques | |||
Très fréquent | douleur musculo-squelettique⁎, arthralgie | douleur musculo-squelettique⁎, arthralgie | arthralgie, douleur musculo-squelettique⁎, myosite⁎, douleur aux extrémités |
Fréquent | myosite⁎, douleur aux extrémités, arthrite⁎ | myosite⁎, douleur aux extrémités, arthrite⁎ | arthrite⁎ |
Peu fréquent | ténosynovite⁎ | ténosynovite⁎ | ténosynovite⁎ |
Rare | syndrome de Sjögren | syndrome de Sjögren | syndrome de Sjögren |
Troubles du rein et des voies urinaires | |||
Fréquent |
| insuffisance rénale aiguë | néphrite⁎ |
Peu fréquent | néphrite⁎ | néphrite⁎, cystite non infectieuse |
|
Rare | cystite non infectieuse |
| cystite non infectieuse |
Troubles généraux et anomalies au site d'administration | |||
Très fréquent | fatigue, asthénie, œdème⁎, fièvre | fatigue, asthénie, fièvre, œdème⁎ | fatigue, asthénie, œdème⁎, fièvre |
Fréquent | syndrome pseudo-grippal, frissons | syndrome pseudo-grippal, frissons | syndrome pseudo-grippal, frissons |
Investigations | |||
Très fréquent |
| augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase | augmentation de la lipase, augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase, augmentation de la créatininémie |
Fréquent | augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase, augmentation des phosphatases alcalines sanguines, hypercalcémie, augmentation de la bilirubinémie, augmentation de la créatininémie | augmentation de la bilirubinémie, augmentation des phosphatases alcalines sanguines, augmentation de la créatininémie, hypercalcémie | augmentation de l’amylase, augmentation de la bilirubinémie, augmentation des phosphatases alcalines sanguines, hypercalcémie |
Peu fréquent | augmentation de l’amylase | augmentation de l’amylase |
|
† Les fréquences des effets indésirables présentées dans le tableau 2 peuvent ne pas être totalement attribuables à pembrolizumab seul mais peuvent aussi intégrer la contribution de la maladie sous-jacente ou des autres médicaments utilisés dans une association.
‡ Sur la base d’une requête standard incluant bradyarythmie et tachyarythmie.
⁎Les termes suivants représentent un groupe d’évènements liés qui décrivent un état pathologique plutôt qu’un évènement isolé :
- anémie hémolytique (anémie hémolytique auto-immune et anémie hémolytique à Coombs négatif)
- réaction liée à la perfusion (hypersensibilité médicamenteuse, réaction anaphylactique, réaction anaphylactoïde, hypersensibilité, réaction d’hypersensibilité liée à la perfusion, syndrome de relargage des cytokines et maladie sérique)
- sarcoïdose (sarcoïdose cutanée et sarcoïdose pulmonaire)
- hypothyroïdie (myxœdème, hypothyroïdie à médiation immunitaire et hypothyroïdie auto-immune)
- insuffisance surrénalienne (maladie d’Addison, insuffisance corticosurrénalienne aiguë, insuffisance corticosurrénalienne secondaire et insuffisance surrénalienne primaire)
- thyroïdite (thyroïdite auto-immune, thyroïdite silencieuse, troubles thyroïdiens, thyroïdite aiguë et thyroïdite à médiation immunitaire)
- hyperthyroïdie (maladie de Basedow)
- hypophysite (hypopituitarisme et hypophysite lymphocytaire)
- diabète de type 1 (acidocétose diabétique)
- syndrome myasthénique (myasthénie grave, y compris exacerbation)
- encéphalite (encéphalite auto-immune et encéphalite non infectieuse)
- syndrome de Guillain-Barré (neuropathie axonale et polyneuropathie démyélinisante)
- myélite (y compris myélite transverse)
- méningite aseptique (méningite et méningite non infectieuse)
- uvéite (choriorétinite, iritis et iridocyclite)
- myocardite (myocardite auto-immune)
- péricardite (péricardite auto-immune, pleuropéricardite et myopéricardite)
- vascularite (vascularite du système nerveux central, aortite et artérite à cellules géantes)
- pneumopathie inflammatoire (pneumopathie interstitielle diffuse, pneumopathie organisée, pneumopathie à médiation immunitaire, maladie pulmonaire à médiation immunitaire et maladie pulmonaire auto-immune)
- douleur abdominale (gêne abdominale, douleur abdominale haute et douleur abdominale basse)
- colite (colite microscopique, entérocolite, entérocolite hémorragique, colite auto-immune et entérocolite à médiation immunitaire)
- gastrite (gastrite érosive, gastrite hémorragique et gastrite à médiation immunitaire)
- pancréatite (pancréatite auto-immune, pancréatite aiguë et pancréatite à médiation immunitaire)
- ulcération gastro-intestinale (ulcère gastrique et ulcère duodénal)
- hépatite (hépatite auto-immune, hépatite à médiation immunitaire, atteinte hépatique d’origine médicamenteuse et hépatite aiguë)
- cholangite sclérosante (cholangite à médiation immunitaire)
- prurit (urticaire, urticaire papuleuse et prurit génital)
- éruption cutanée (éruption érythémateuse, éruption folliculaire, éruption maculaire, éruption maculo-papuleuse, éruption papuleuse, éruption pruritigineuse, éruption vésiculaire et rash génital)
- réactions cutanées sévères (rash exfoliatif, pemphigus, et évènements suivants de Grade ≥ 3 : vascularite cutanée, dermatite bulleuse, dermatite exfoliative, dermatite exfoliative généralisée, érythème polymorphe, lichen plan, lichen plan buccal, pemphigoïde, prurit, prurit génital, éruption cutanée, éruption cutanée érythémateuse, éruption maculo-papuleuse, éruption cutanée prurigineuse, éruption pustuleuse, nécrose cutanée et éruption cutanée toxique)
- vitiligo (dépigmentation cutanée, hypopigmentation cutanée et hypopigmentation de la paupière)
- kératose lichénoïde (lichen plan et lichen scléreux)
- douleur musculo-squelettique (gêne musculo-squelettique, douleur dorsale, raideur musculo-squelettique, douleur thoracique musculo-squelettique et torticolis)
- myosite (myalgie, myopathie, myosite nécrosante, pseudo-polyarthrite rhizomélique et rhabdomyolyse)
- arthrite (gonflement des articulations, polyarthrite, épanchement articulaire, arthrite auto-immune et arthrite à médiation immunitaire)
- ténosynovite (tendinite, synovite et douleur aux tendons)
- néphrite (néphrite auto-immune, néphrite à médiation immunitaire, néphrite tubulo-interstitielle et insuffisance rénale, insuffisance rénale aiguë ou atteinte rénale aiguë avec néphrite avérée, syndrome néphrotique, glomérulonéphrite, glomérulonéphrite membraneuse et glomérulonéphrite aiguë)
- œdème (œdème périphérique, œdème généralisé, surcharge liquidienne, rétention liquidienne, œdème palpébral et œdème labial, œdème du visage, œdème localisé et œdème périorbitaire)
Pembrolizumab en association à l'enfortumab vedotin (voir rubrique 4.2)
Lorsque pembrolizumab est administré en association à l'enfortumab vedotin, se référer au RCP de l'enfortumab vedotin avant l'initiation du traitement.
La sécurité de pembrolizumab en association à l'enfortumab vedotin a été évaluée chez 564 patients atteints d'un carcinome urothélial non résécable ou métastatique recevant 200 mg de pembrolizumab le Jour 1 et 1,25 mg/kg d'enfortumab vedotin les Jours 1 et 8 de chaque cycle de 21 jours.
Dans l'ensemble, l'incidence des effets indésirables de pembrolizumab en association à l'enfortumab vedotin a été observée comme étant plus élevée que celle de pembrolizumab en monothérapie, reflétant la contribution de l'enfortumab vedotin et la durée du traitement en association plus longue.
Les effets indésirables étaient généralement similaires à ceux observés chez les patients recevant pembrolizumab ou enfortumab vedotin en monothérapie. L'incidence de l’éruption cutanée maculo-papuleuse était de 36 % tous Grades (10 % de Grades 3-4), ce qui est plus élevé que celle observée avec pembrolizumab en monothérapie.
Généralement similaires aux observations avec le comparateur chimiothérapie, les fréquences des événements indésirables étaient plus élevées chez les patients âgés de ≥ 65 ans comparées aux patients âgés de < 65 ans, en particulier pour les événements indésirables graves (respectivement 56,3 % et 35,3 %) et les événements de Grade ≥ 3 (respectivement 80,3 % et 64,2 %).
Description d’une sélection d’effets indésirables
Les données concernant les effets indésirables à médiation immunitaire suivants sont basées sur les patients ayant reçu pembrolizumab selon quatre posologies (2 mg/kg de poids corporel toutes les 3 semaines, 10 mg/kg de poids corporel toutes les 2 ou 3 semaines ou 200 mg toutes les 3 semaines) dans les études cliniques (voir rubrique 5.1). Les recommandations de prise en charge de ces effets indésirables sont décrites en rubrique 4.4.
Effets indésirables à médiation immunitaire (voir rubrique 4.4)
Pneumopathie inflammatoire à médiation immunitaire
Une pneumopathie inflammatoire est survenue chez 324 (4,2 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3, 4 ou 5 chez 143 (1,9 %), 81 (1,1 %), 19 (0,2 %) et 9 (0,1 %) patients, respectivement. Le délai d’apparition médian d’une pneumopathie inflammatoire a été de 3,9 mois (de 2 jours à 27,2 mois) et la durée médiane a été de 2,0 mois (de 1 jour à 51,0+ mois). La pneumopathie inflammatoire était plus fréquente chez les patients ayant des antécédents d’irradiation thoracique antérieure (8,1 %) que chez les patients n'ayant pas reçu d’irradiation thoracique préalable (3,9 %). Une pneumopathie inflammatoire a conduit à un arrêt de pembrolizumab chez 131 (1,7 %) patients. La pneumopathie inflammatoire s’est résolue chez 196 patients, 6 avec des séquelles.
Pour les patients atteints de CBNPC, une pneumopathie inflammatoire est survenue chez 230 (6,1 %), y compris des cas de Grade 2, 3, 4 ou 5 chez 103 (2,7 %), 63 (1,7 %), 17 (0,4 %) et 10 (0,3 %) patients, respectivement. Chez les patients atteints de CBNPC localement avancé ou métastatique, une pneumopathie inflammatoire est survenue chez 8,9 % patients ayant des antécédents d’irradiation thoracique antérieure. Chez les patients atteints de LHc, l’incidence de la pneumopathie inflammatoire (tous grades) variait entre 5,2 % et 10,8 % pour les patients avec un LHc dans l’étude KEYNOTE-087 (n = 210) et l’étude KEYNOTE-204 (n = 148), respectivement.
Colite à médiation immunitaire
Une colite est survenue chez 158 (2,1 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 49 (0,6 %), 82 (1,1 %) et 6 (0,1 %) patients, respectivement. Le délai d’apparition médian de la colite a été de 4,3 mois (de 2 jours à 24,3 mois) et la durée médiane a été de 1,1 mois (de 1 jour à 45,2 mois). Une colite a conduit à un arrêt de pembrolizumab chez 48 (0,6 %) patients. La colite s’est résolue chez 132 patients, 2 avec des séquelles. Chez les patients atteints de cancer colorectal traités par pembrolizumab en monothérapie (n = 153), l’incidence de la colite était de 6,5 % (tous grades) avec 2,0 % de Grade 3 et 1,3 % de Grade 4.
Hépatite à médiation immunitaire
Une hépatite est survenue chez 80 (1,0 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 12 (0,2 %), 55 (0,7 %) et 8 (0,1 %) patients, respectivement. Le délai d’apparition médian de l’hépatite a été de 3,5 mois (de 8 jours à 26,3 mois) et la durée médiane a été de 1,3 mois (de 1 jour à 29,0+ mois). Une hépatite a conduit à un arrêt de pembrolizumab chez 37 (0,5 %) patients. L’hépatite s’est résolue chez 60 patients.
Néphrite à médiation immunitaire
Une néphrite est survenue chez 37 (0,5 %) patients recevant pembrolizumab en monothérapie, y compris des cas de Grade 2, 3 ou 4 chez 11 (0,1 %), 19 (0,2 %) et 2 (< 0,1 %) patients, respectivement. Le délai d’apparition médian de la néphrite a été de 4,2 mois (de 12 jours à 21,4 mois) et la durée médiane a été de 3,3 mois (de 6 jours à 28,2+ mois). Une néphrite a conduit à un arrêt de pembrolizumab chez 17 (0,2 %) patients. La néphrite s’est résolue chez 25 patients, 5 avec des séquelles. Chez les patients atteints d’un CBNPC non-épidermoïde traités par pembrolizumab associé à une chimiothérapie pemetrexed et sel de platine (n = 488), l’incidence de la néphrite était de 1,4 % (tous grades) avec 0,8 % de Grade 3 et 0,4 % de Grade 4.
Endocrinopathies à médiation immunitaire
Une insuffisance surrénalienne est survenue chez 74 (1,0 %) patients, y compris des cas de Grade 2, 3 et 4 chez 34 (0,4 %), 31 (0,4 %) et 4 (0,1 %) patients recevant pembrolizumab, respectivement. Le délai d’apparition médian de l’insuffisance surrénalienne a été de 5,4 mois (de 1 jour à 23,7 mois) et la durée médiane n’a pas été atteinte (de 3 jours à 40,1+ mois). Une insuffisance surrénalienne a conduit à un arrêt de pembrolizumab chez 13 (0,2 %) patients. L’insuffisance surrénalienne s’est résolue chez 28 patients, 11 avec des séquelles.
Une hypophysite est survenue chez 52 (0,7 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 23 (0,3 %), 24 (0,3 %) et 1 (< 0,1 %) patients, respectivement. Le délai d’apparition médian de l’hypophysite a été de 5,9 mois (de 1 jour à 17,7 mois) et la durée médiane a été de 3,6 mois (de 3 jours à 48,1+ mois). Une hypophysite a conduit à un arrêt de pembrolizumab chez 14 (0,2 %) patients. L’hypophysite s’est résolue chez 23 patients, 8 avec des séquelles.
Une hyperthyroïdie est survenue chez 394 (5,2 %) patients recevant pembrolizumab, y compris des cas de Grade 2 ou 3 chez 108 (1,4 %) et 9 (0,1 %) patients, respectivement. Le délai d’apparition médian de l’hyperthyroïdie a été de 1,4 mois (de 1 jour à 23,2 mois) et la durée médiane a été de 1,6 mois (de 4 jours à 43,1+ mois). Une hyperthyroïdie a conduit à un arrêt de pembrolizumab chez 4 (0,1 %) patients. L’hyperthyroïdie s’est résolue chez 326 (82,7 %) patients, 11 avec des séquelles. Chez les patients atteints d’un mélanome, d’un CBNPC et d’un CCR traités par pembrolizumab en monothérapie au stade adjuvant (n = 2 060), l'incidence de l'hyperthyroïdie était de 11,0 %, dont la majorité était de Grade 1 ou 2.
Une hypothyroïdie est survenue chez 939 (12,3 %) patients recevant pembrolizumab, y compris des cas de Grade 2 ou 3 chez 687 (9,0 %) et 8 (0,1 %) patients, respectivement. Le délai d’apparition médian de l’hypothyroïdie a été de 3,4 mois (de 1 jour à 25,9 mois) et la durée médiane n’a pas été atteinte (de 2 jours à 63,0+ mois). L’hypothyroïdie a conduit à un arrêt de pembrolizumab chez 6 (0,1 %) patients. L’hypothyroïdie s’est résolue chez 216 (23,0 %) patients, 16 avec des séquelles. Chez les patients atteints d’un LHc (n = 389), l’incidence de l’hypothyroïdie était de 17 %, toutes étant de Grade 1 ou 2. Chez les patients atteints d’un CETEC récidivant ou métastatique traités par pembrolizumab en monothérapie (n = 909), l’incidence de l’hypothyroïdie était de 16,1 % (tous grades) avec 0,3 % de Grade 3. Chez les patients atteints d’un CETEC récidivant ou métastatique traités par pembrolizumab en association à une chimiothérapie à base de sels de platine et de 5-FU (n = 276), l’incidence de l’hypothyroïdie était de 15,2 %, toutes étant de Grade 1 ou 2. Chez les patients atteints d’un CETEC localement avancé résécable traités par pembrolizumab en traitement néoadjuvant et en association à une radiothérapie avec ou sans cisplatine concomitant en traitement adjuvant (n = 361), l’incidence de l’hypothyroïdie était de 24,7 %, toutes étant de Grade 1 ou 2. Chez les patients traités par pembrolizumab en association à l’axitinib ou au lenvatinib (n = 1 456), l'incidence de l'hypothyroïdie était de 46,2 % (tous Grades) avec 0,8 % de Grade 3 ou 4. Chez les patients atteints d’un mélanome, d’un CBNPC et d’un CCR traités par pembrolizumab en monothérapie au stade adjuvant (n = 2 060), l'incidence de l'hypothyroïdie était de 18,5 %, dont la majorité était de Grade 1 ou 2.
Effets indésirables cutanés à médiation immunitaire
Des réactions cutanées sévères à médiation immunitaire sont survenues chez 130 (1,7 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3, 4 ou 5 chez 11 (0,1 %), 103 (1,3 %), 1 (< 0,1 %) et 1 (< 0,1%) patients, respectivement. Le délai d’apparition médian des réactions cutanées sévères a été de 2,8 mois (de 2 jours à 25,5 mois). La durée médiane a été de 1,9 mois (de 1 jour à 47,1+ mois). Des réactions cutanées sévères ont conduit à un arrêt de pembrolizumab chez 18 (0,2 %) patients. Les réactions cutanées sévères se sont résolues chez 95 patients, 2 avec des séquelles.
De rares cas de SSJ et de NET, dont certains d’issue fatale, ont été observés (voir rubriques 4.2 et 4.4).
Complications d’une GCSH allogénique dans le LHc
Parmi les 14 patients de KEYNOTE-013 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 6 patients ont développé une GVH aiguë et 1 patient a développé une GVH chronique, dont aucune n’a été fatale. Deux patients ont présenté une MVO hépatique, dont une d’issue fatale. Un patient a présenté un syndrome de prise de greffe après transplantation.
Parmi les 32 patients de KEYNOTE-087 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 16 patients ont développé une GVH aiguë et 7 patients ont développé une GVH chronique, dont deux d’issue fatale. Aucun patient n’a présenté de MVO hépatique. Aucun patient n’a présenté de syndrome de prise de greffe après transplantation.
Parmi les 14 patients de KEYNOTE-204 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 8 patients ont développé une GVH aiguë et 3 patients ont développé une GVH chronique, aucune d’issue fatale. Aucun patient n’a présenté de MVO hépatique. Un patient a présenté un syndrome de prise de greffe après transplantation.
Enzymes hépatiques élevées lorsque pembrolizumab est associé à l’axitinib dans le CCR
Dans une étude clinique chez des patients atteints d’un CCR non préalablement traité recevant pembrolizumab en association à l’axitinib, une augmentation des ALAT (20 %) et des ASAT (13 %) de Grades 3 et 4 a été observée avec une incidence plus élevée qu’attendue. La durée médiane d’apparition de l’augmentation des ALAT était de 2,3 mois (de 7 jours à 19,8 mois). Chez les patients avec des ALAT ≥ 3 fois la LSN (Grades 2-4, n = 116), l’augmentation des ALAT s’est améliorée jusqu’aux Grades 0-1 chez 94 % d’entre eux. Cinquante-neuf pour cent des patients présentant des ALAT augmentées ont reçu des corticostéroïdes systémiques. Parmi les patients qui se sont rétablis, une réintroduction a été faite chez 92 (84 %) d’entre eux avec soit pembrolizumab (3 %) soit axitinib (31 %) en monothérapie soit les deux (50 %). Parmi ces patients, 55 % n’ont pas eu de réapparition des ALAT > 3 fois la LSN, et parmi les patients ayant présenté une réapparition des ALAT > 3 fois la LSN, tous se sont rétablis. Il n’y a pas eu d’effets indésirables hépatiques de Grade 5.
Anomalies des valeurs biologiques
Chez les patients traités par pembrolizumab en monothérapie, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 9,9 % pour une diminution des lymphocytes, 7,3 % pour une diminution du sodium, 5,7 % pour une diminution de l’hémoglobine, 4,6 % pour une augmentation du glucose, 4,5 % pour une diminution du phosphate, 3,1 % pour une augmentation des ALAT, 2,9 % pour une augmentation des ASAT, 2,6 % pour une augmentation des phosphatases alcalines, 2,2 % pour une diminution du potassium, 2,1 % pour une diminution des neutrophiles, 1,7 % pour une augmentation de la bilirubine, 1,7 % pour une diminution des plaquettes, 1,7 % pour une augmentation du potassium, 1,6 % pour une augmentation du calcium, 1,4 % pour une diminution de l’albumine, 1,3 % pour une diminution du calcium, 1,2 % pour une augmentation de la créatinine, 0,8 % pour une diminution des leucocytes, 0,8 % pour une augmentation du magnésium, 0,6 % pour une diminution du glucose, 0,2 % pour une diminution du magnésium et 0,2 % pour une augmentation du sodium.
Chez les patients traités par pembrolizumab en association à la chimiothérapie, à la RT ou à la RCT, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 36,2 % pour une diminution des neutrophiles, 31,9 % pour une diminution des lymphocytes, 23,7 % pour une diminution des leucocytes, 20,3 % pour une diminution de l’hémoglobine, 11,8 % pour une diminution des plaquettes, 9,6 % pour une diminution du sodium, 7,8 % pour une diminution du potassium, 7,2 % pour une diminution du phosphate, 5,5 % pour une augmentation du glucose, 5,2 % pour une augmentation des ALAT, 4,6 % pour une augmentation des ASAT, 3,4 % pour une diminution du calcium, 3,0 % pour une augmentation de la bilirubine, 3,0 % pour une augmentation du potassium, 2,9 % pour une augmentation de la créatinine, 2,4 % pour une augmentation des phosphatases alcalines, 2,2 % pour une diminution de l’albumine, 1,6 % pour une augmentation du calcium, 0,8 % pour une diminution du glucose et 0,4 % pour une augmentation du sodium.
Chez les patients traités par pembrolizumab en association à l’axitinib ou au lenvatinib, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 23,0 % pour une augmentation de la lipase (non mesurée chez les patients traités par pembrolizumab et axitinib), 12,3 % pour une diminution des lymphocytes, 11,4 % pour une diminution du sodium, 11,2 % pour une augmentation de l'amylase, 11,2 % pour une augmentation des triglycérides, 10,4 % pour une augmentation des ALAT, 8,9 % pour une augmentation des ASAT, 7,8 % pour une augmentation du glucose, 6,8 % pour une diminution du phosphate, 6,1 % pour une diminution du potassium, 5,1 % pour une augmentation du potassium, 4,5 % pour une augmentation du cholestérol, 4,4 % pour une augmentation de la créatinine, 4,2 % pour une diminution de l'hémoglobine, 4,0 % pour une diminution des neutrophiles, 3,1 % pour une augmentation des phosphatases alcalines, 3,0 % pour une diminution des plaquettes, 2,8 % pour une augmentation de la bilirubine, 2,2 % pour une diminution du calcium, 2,2 % pour une augmentation du magnésium, 1,7 % pour une diminution des leucocytes, 1,5 % pour une diminution du magnésium, 1,5 % pour une augmentation de l’INR (prothrombine), 1,4 % pour une diminution du glucose, 1,2 % pour une diminution de l'albumine, 1,0 % pour une augmentation du calcium, 0,4 % pour une augmentation du sodium et 0,1 % pour une augmentation de l'hémoglobine.
Immunogénicité
Dans les études cliniques menées chez les patients traités par pembrolizumab en monothérapie à la dose de 2 mg/kg de poids corporel toutes les trois semaines, 200 mg toutes les trois semaines ou 10 mg/kg de poids corporel toutes les 2 ou 3 semaines, 36 (1,8 %) des 2 034 patients évaluables ont été testés positifs pour des anticorps anti-pembrolizumab apparus au cours du traitement dont 9 (0,4 %) patients avec des anticorps neutralisants contre pembrolizumab. Il n’a pas été mis en évidence de modification du profil pharmacocinétique ou de tolérance en présence d’anticorps anti-pembrolizumab liants ou neutralisants.
Population pédiatrique
La sécurité de pembrolizumab en monothérapie a été évaluée à la dose de 2 mg/kg de poids corporel toutes les 3 semaines dans l’étude de phase I/II KEYNOTE-051 réalisée chez 161 patients pédiatriques âgés de 9 mois à 17 ans atteints d’un mélanome avancé, d’un lymphome ou de tumeurs solides avancées PD-L1 positives, en rechute ou réfractaires. La population avec un LHc (n = 22) incluait des patients âgés de 11 à 17 ans. Le profil de sécurité chez les patients pédiatriques était généralement similaire à celui observé chez les adultes traités par pembrolizumab. Les effets indésirables les plus fréquents (rapportés chez au moins 20 % des patients pédiatriques) étaient : pyrexie (33 %), vomissements (30 %), céphalées (26 %), douleurs abdominales (22 %), anémie (21 %), toux (21 %) et constipation (20 %). La majorité des effets indésirables rapportés en monothérapie étaient d’une sévérité de Grades 1 ou 2. Soixante-seize (47,2 %) patients présentaient 1 ou plusieurs effets indésirables de Grades 3 à 5 dont 5 (3,1 %) patients avec 1 ou plusieurs effets indésirables ayant entraîné le décès. Les fréquences sont basées sur tous les effets indésirables rapportés, quelle que soit l’évaluation de la causalité par l’investigateur. Les données de sécurité à long terme du pembrolizumab chez les adolescents atteints de mélanome de stade IIB, IIC et III traités au stade adjuvant sont actuellement indisponibles.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration: en Belgique : Agence Fédérale des Médicaments et des Produits de Santé, www.afmps.be - Division Vigilance : Site internet: www.notifieruneffetindesirable.be, e-mail: adr@fagg-afmps.be, au Luxembourg : Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet: www.guichet.lu/pharmacovigilance.
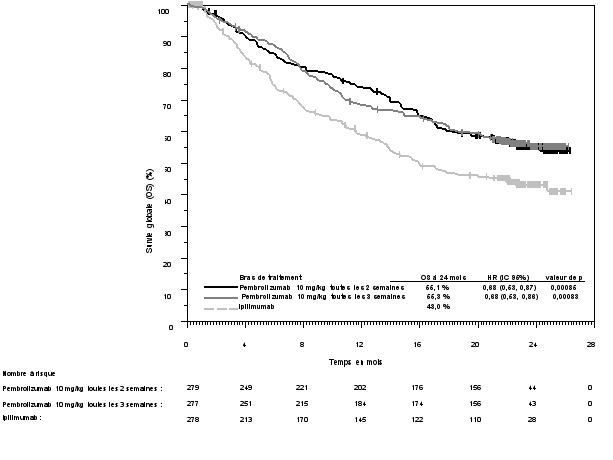
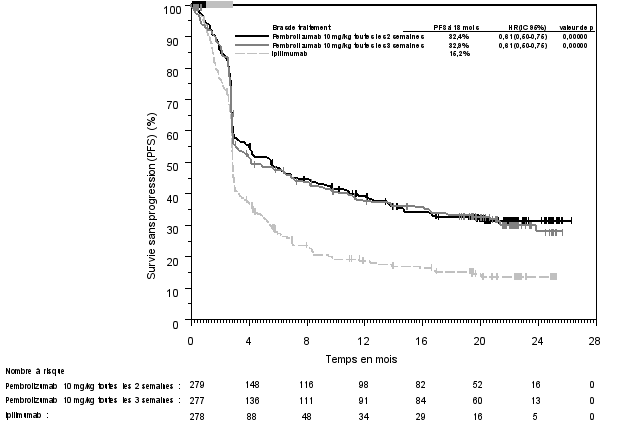
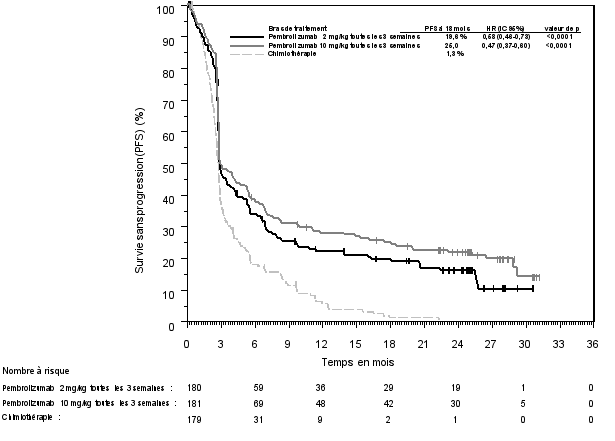

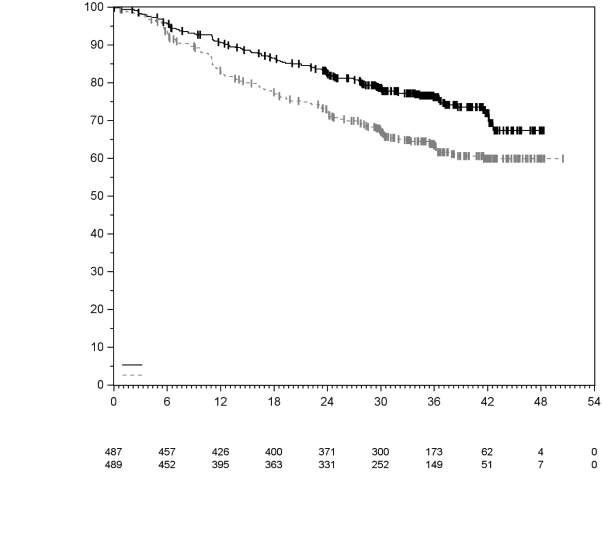






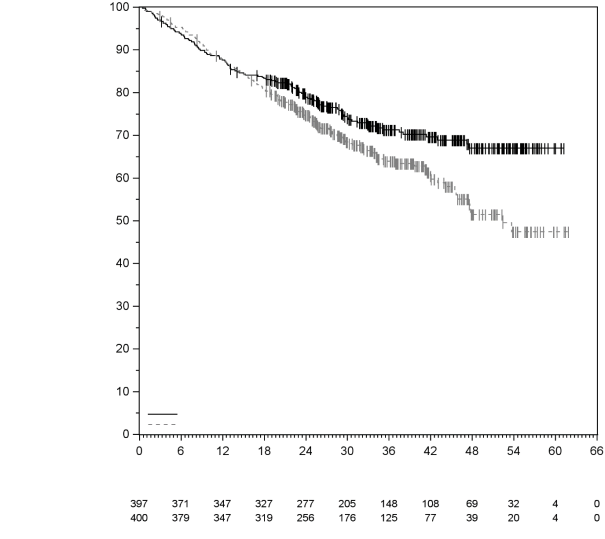


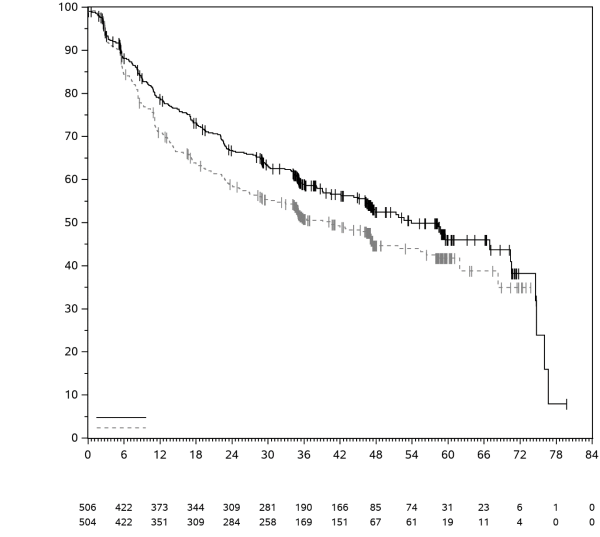
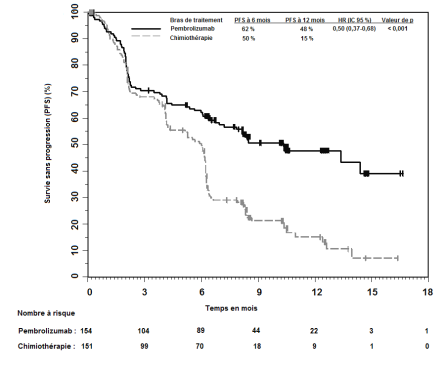
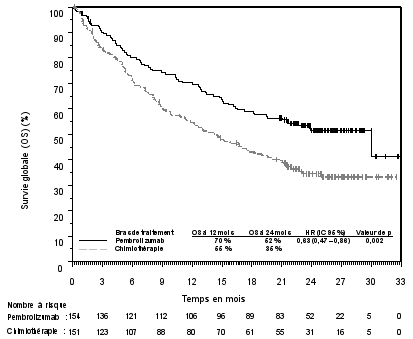

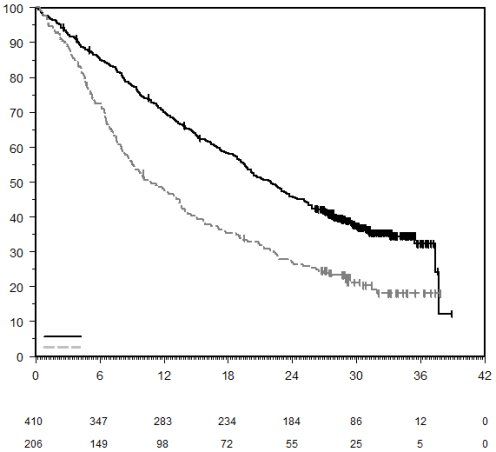


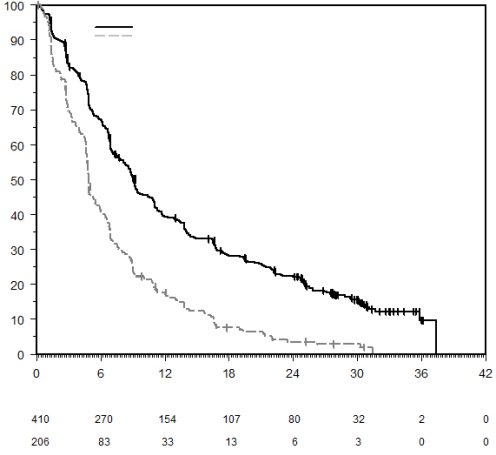
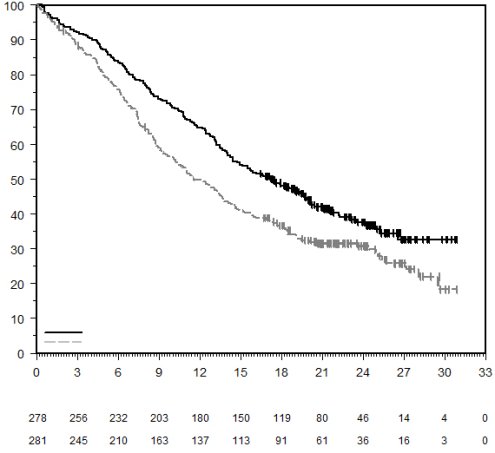

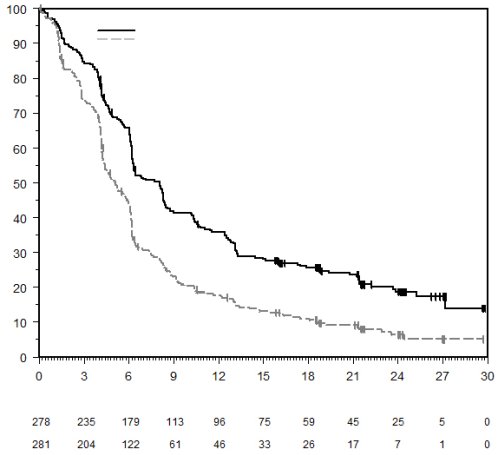







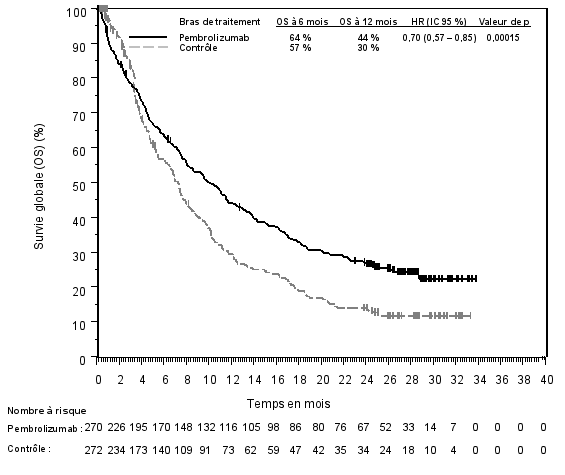
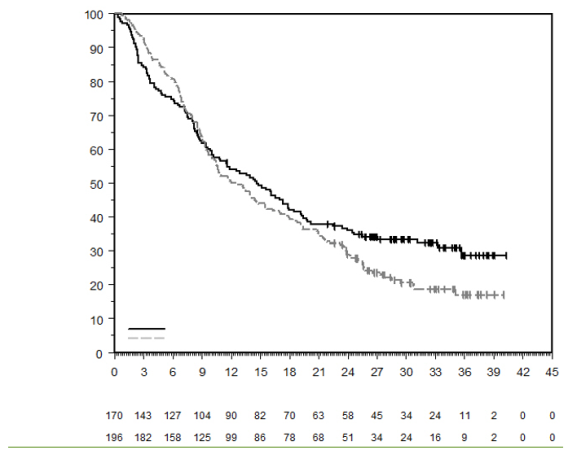
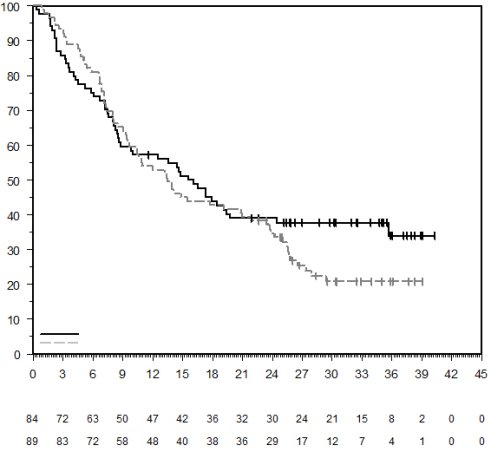
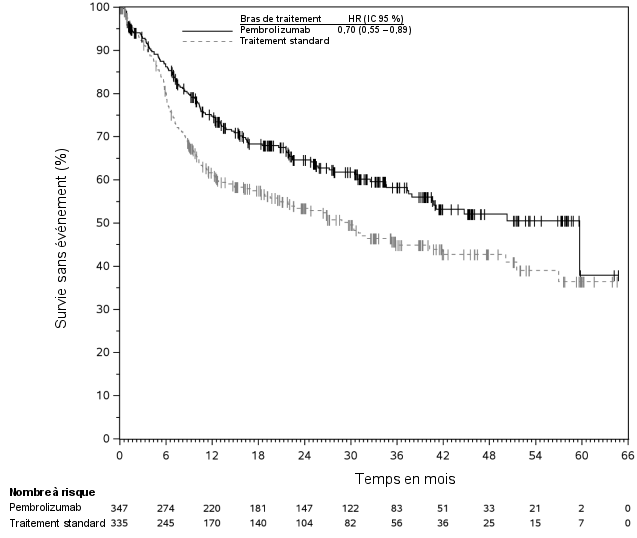
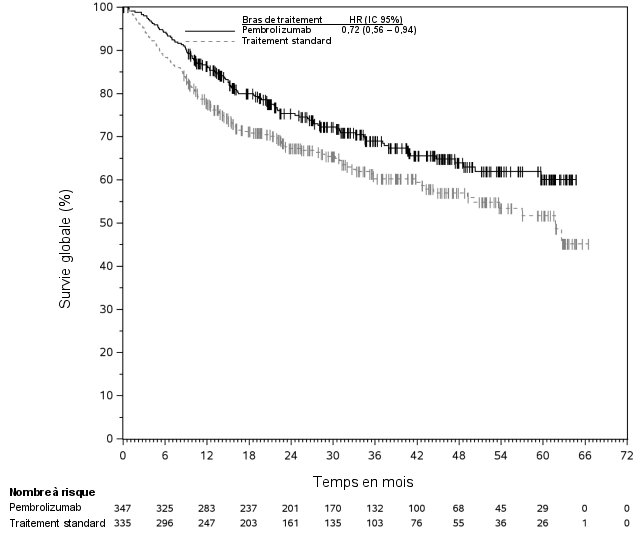
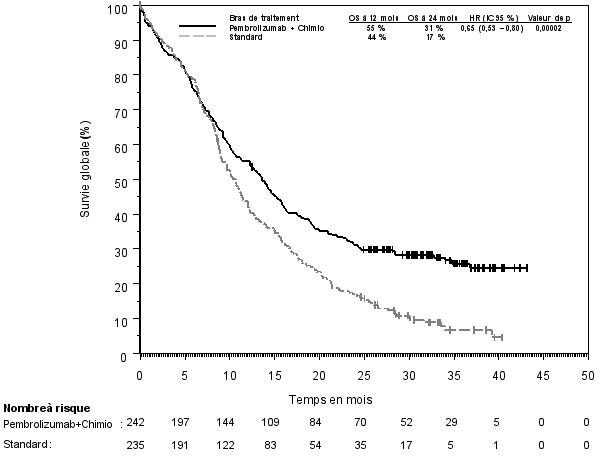
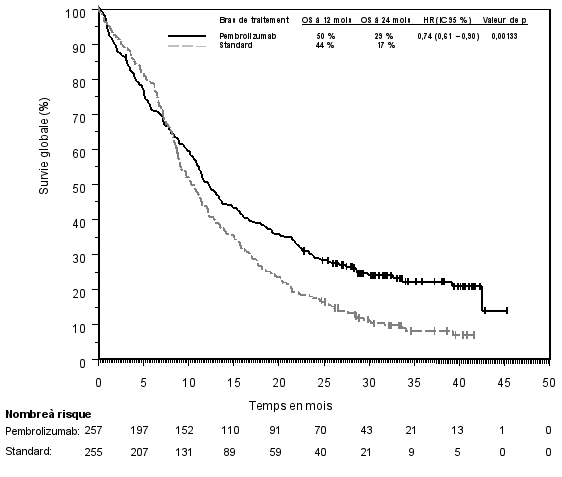
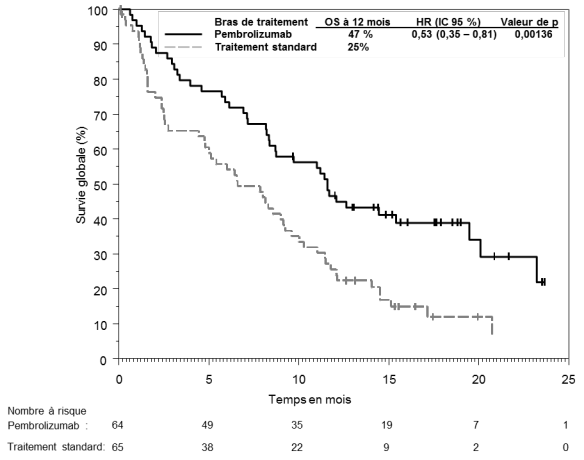






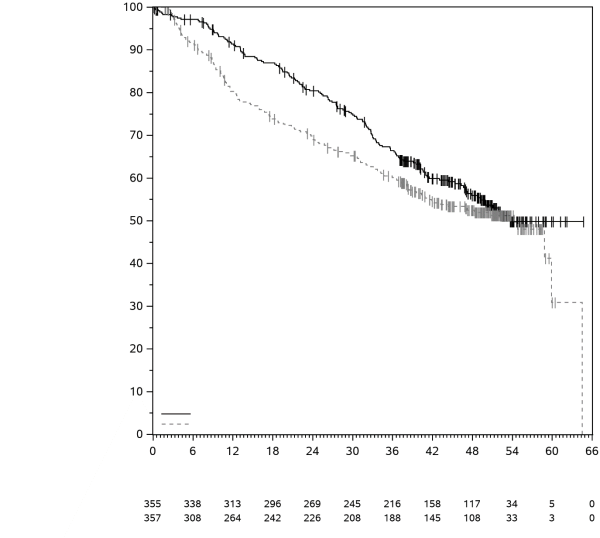
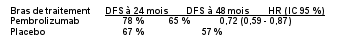

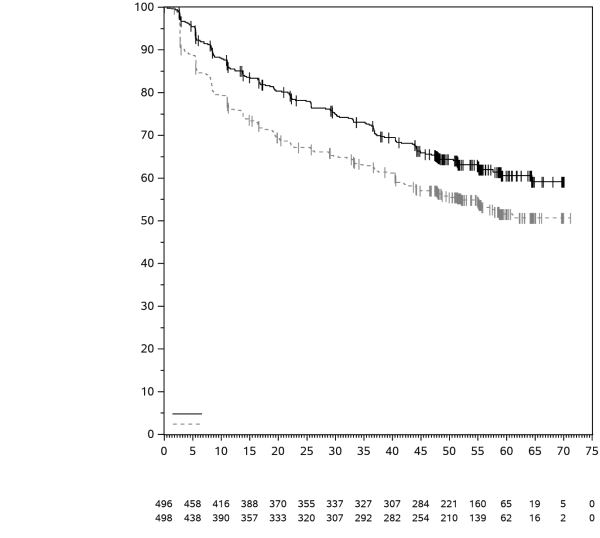



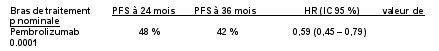

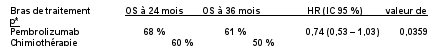

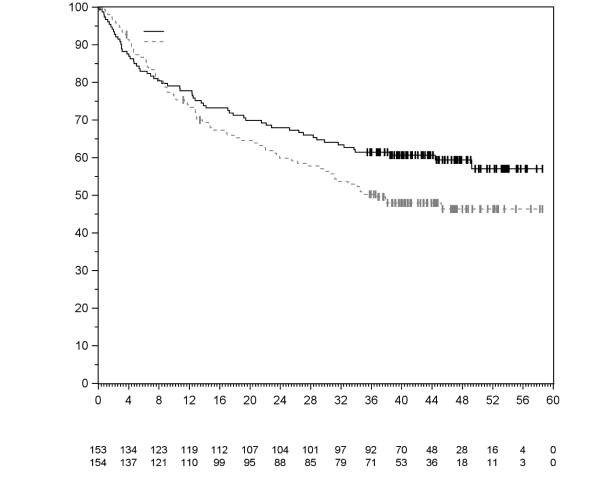







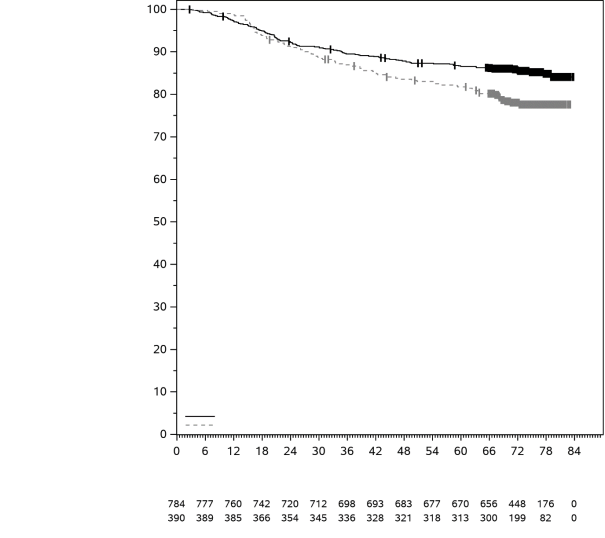



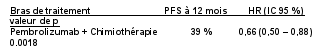
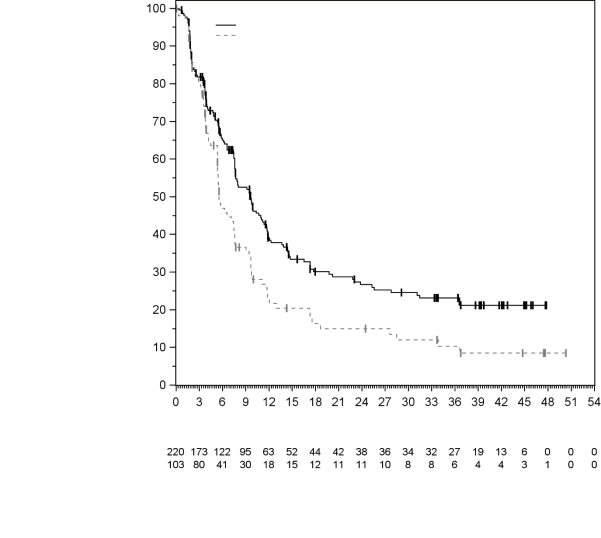
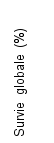


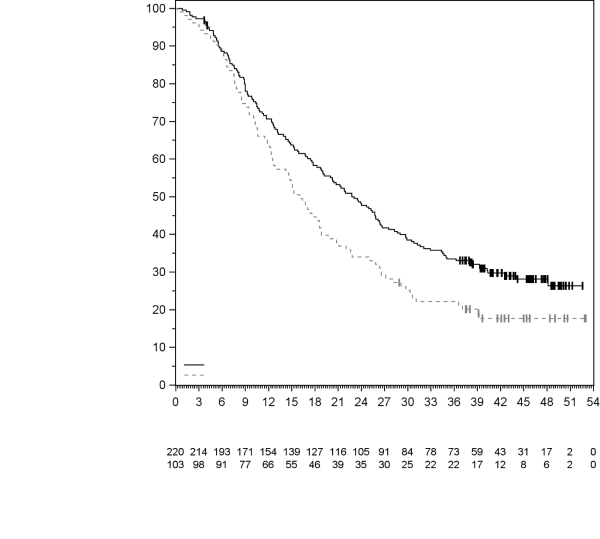



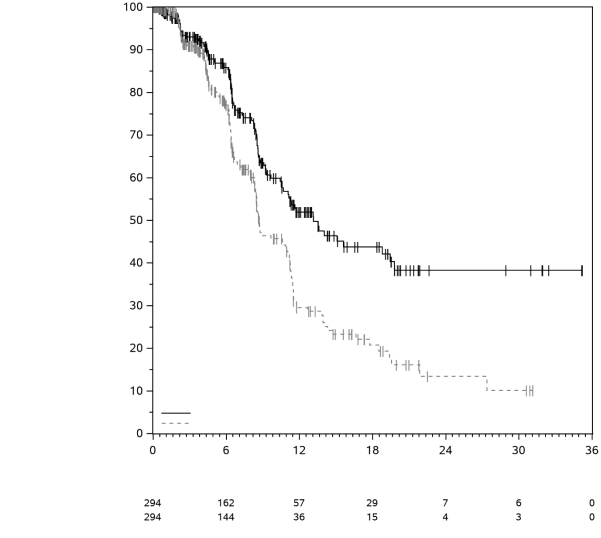

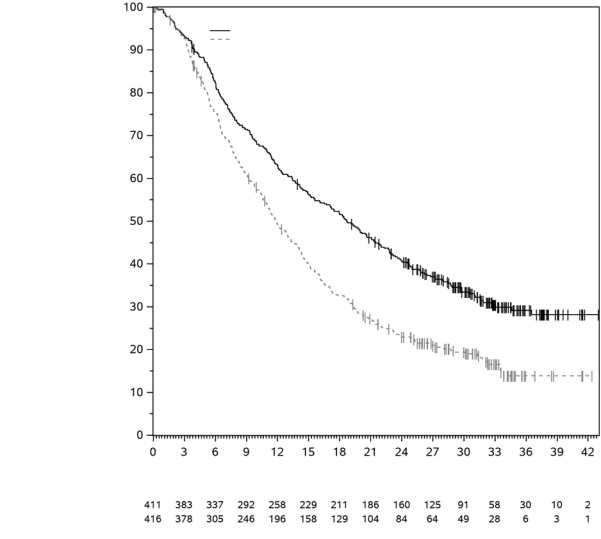
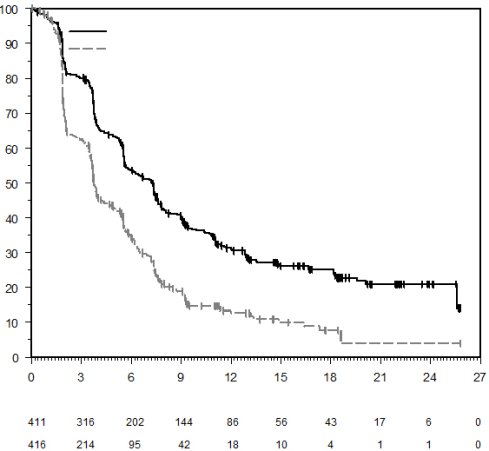
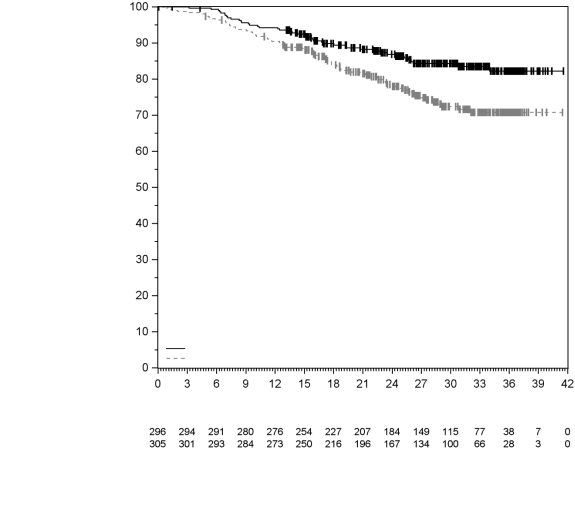



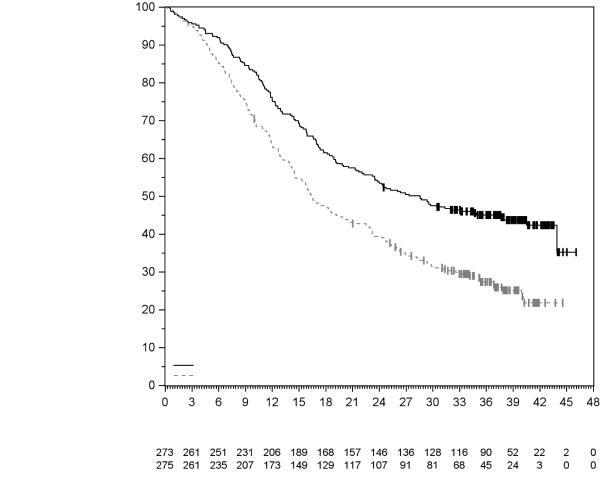

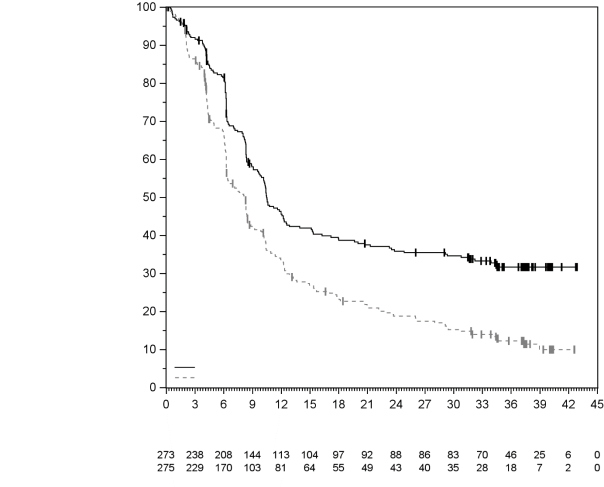


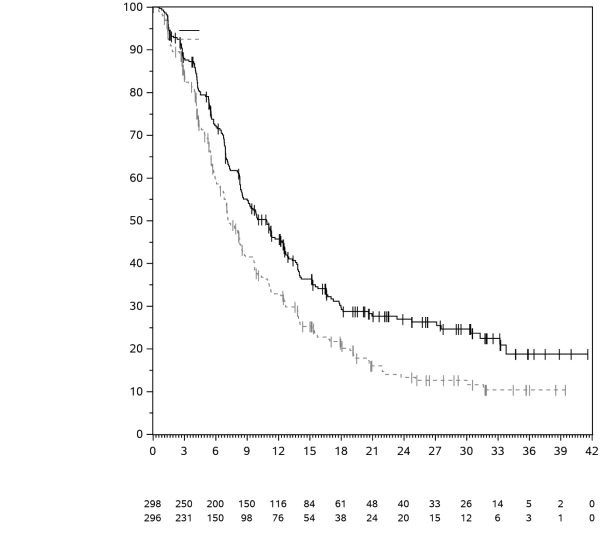

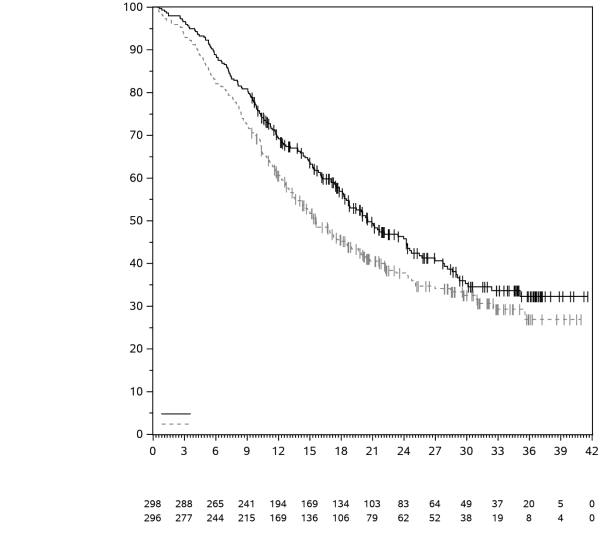










7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
Merck Sharp & Dohme B.V.
Waarderweg 39
2031 BN Haarlem
Pays-Bas
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/15/1024/002
EU/1/15/1024/003
10. DATE DE MISE À JOUR DU TEXTE
11/1025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence européenne des médicaments https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 3543287 | KEYTRUDA 25MG/ML CONC POUR SOL PERF VIAL 1 X 4ML | L01FF02 | - | € 2647,1 | Oui | - | - |