RÉSUMÉ DES CARACTÉRISTIQUES DU PRODUIT
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
1. DÉNOMINATION DU MÉDICAMENT
Benlysta 120 mg poudre pour solution à diluer pour perfusion.
Benlysta 400 mg poudre pour solution à diluer pour perfusion.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Benlysta 120 mg poudre pour solution à diluer pour perfusion.
Chaque flacon contient 120 mg de bélimumab. Après reconstitution, la solution contient 80 mg de bélimumab par mL.
Benlysta 400 mg poudre pour solution à diluer pour perfusion.
Chaque flacon contient 400 mg de bélimumab. Après reconstitution, la solution contient 80 mg de bélimumab par mL.
Le bélimumab est un anticorps monoclonal humain de type IgG1λ, produit dans une lignée cellulaire de mammifères (NS0) par la technique de l’ADN recombinant.
Excipient à effet notoire
Benlysta 120 mg poudre pour solution à diluer pour perfusion.
Chaque flacon contient 0,6 mg de polysorbate 80.
Benlysta 400 mg poudre pour solution à diluer pour perfusion.
Chaque flacon contient 2,0 mg de polysorbate 80.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Poudre pour solution à diluer pour perfusion.
Poudre blanche à blanc cassé.
4. INFORMATIONS CLINIQUES
4.1 Indications thérapeutiques
Benlysta, en association au traitement habituel, est indiqué chez les patients âgés de 5 ans et plus atteints de lupus systémique (LS) actif avec présence d’auto-anticorps et activité de la maladie élevée (définie par exemple par la présence d’anticorps anti-ADN natif et un complément bas) malgré un traitement standard (voir rubrique 5.1).
Benlysta est indiqué en association avec des immunosuppresseurs pour le traitement des patients adultes atteints de glomérulonéphrite lupique active (voir rubriques 4.2 et 5.1).
4.2 Posologie et mode d’administration
Le traitement par Benlysta doit être instauré et surveillé par un médecin expérimenté dans le diagnostic et le traitement du lupus systémique. Les perfusions de Benlysta doivent être administrées par un professionnel de santé qualifié et formé à l’administration d’un traitement par perfusion.
L’administration de Benlysta peut provoquer des réactions d’hypersensibilité et des réactions liées à la perfusion, sévères ou mettant en jeu le pronostic vital. La survenue de symptômes d'hypersensibilité aiguë a été notifiée chez des patients plusieurs heures après l’administration de la perfusion. Une réapparition de réactions cliniquement significatives après un traitement initial approprié des symptômes a également été observée (voir rubriques 4.4 et 4.8). Par conséquent, Benlysta doit être administré dans un environnement disposant des moyens nécessaires pour prendre en charge immédiatement ce type de réactions. Il est recommandé que les patients restent sous surveillance médicale pendant une période prolongée (plusieurs heures), après les 2 premières perfusions au minimum compte tenu de la possibilité d'une réaction retardée.
Les patients traités par Benlysta doivent être informés du risque potentiel d’hypersensibilité sévère ou mettant en jeu le pronostic vital et de la possibilité d’une apparition retardée ou d’une récurrence des symptômes. La notice devra être fournie au patient à chaque administration de Benlysta (voir rubrique 4.4).
Posologie
Une prémédication avec un anti-histaminique, avec ou sans antipyrétique, peut être administrée avant la perfusion de Benlysta (voir rubrique 4.4).
Chez les patients atteints de LS ou de glomérulonéphrite lupique active, la posologie recommandée de Benlysta est de 10 mg/kg de poids corporel aux jours 0, 14 et 28 du traitement, puis toutes les 4 semaines. L'état du patient doit être régulièrement évalué.
Chez les patients atteints de LS, l’arrêt du traitement par Benlysta doit être envisagé en l’absence d’amélioration du contrôle de la maladie après 6 mois de traitement.
Chez les patients atteints de glomérulonéphrite lupique active, Benlysta doit être utilisé en association avec des corticoïdes et du mycophénolate ou du cyclophosphamide pour l’induction, ou du mycophénolate ou de l’azathioprine pour l’entretien.
Passage de la voie intraveineuse à la voie sous-cutanée
Lupus systémique (LS)
Si un patient atteint de LS passe d’un traitement par Benlysta administré par voie intraveineuse à un traitement par Benlysta administré par voie sous-cutanée, la première injection par voie sous-cutanée doit être réalisée 1 à 4 semaines après la dernière dose par voie intraveineuse (voir rubrique 5.2).
Glomérulonéphrite lupique
Si un patient atteint de glomérulonéphrite lupique passe d’un traitement par Benlysta administré par voie intraveineuse à un traitement par Benlysta administré par voie sous-cutanée, il est recommandé que la première dose de 200 mg injectée par voie sous-cutanée soit réalisée 1 à 2 semaines après la dernière dose administrée par voie intraveineuse. Cette transition peut avoir lieu à tout moment après que le patient ait reçu les deux premières administrations par voie intraveineuse (voir rubrique 5.2).
Populations spéciales
Sujets âgés
Les données chez les sujets ≥ 65 ans sont limitées (voir rubrique 5.1). Benlysta doit être utilisé avec prudence chez les sujets âgés. Aucun ajustement posologique n’est requis (voir rubrique 5.2).
Insuffisance rénale
Le bélimumab a été évalué chez un nombre limité de patients ayant un lupus systémique avec une insuffisance rénale.
Sur la base des informations disponibles, aucun ajustement posologique n’est requis chez les patients ayant une insuffisance rénale légère, modérée ou sévère. Cependant, la prudence est recommandée chez les patients ayant une insuffisance rénale sévère en raison de l’absence de données dans cette population (voir rubrique 5.2).
Insuffisance hépatique
Aucune étude spécifique n’a été menée avec Benlysta chez des patients ayant une insuffisance hépatique. Il est peu probable qu’un ajustement de la posologie soit nécessaire chez ces patients (voir rubrique 5.2).
Population pédiatrique
Lupus systémique (LS)
La posologie recommandée de Benlysta pour les enfants âgés de 5 ans et plus, est de 10 mg/kg de poids corporel aux jours 0, 14 et 28 du traitement, puis toutes les 4 semaines.
La sécurité et l’efficacité de Benlysta par voie intraveineuse chez les enfants de moins de 5 ans et de moins de 15 kg n’ont pas été évaluées. Aucune donnée n’est disponible.
Glomérulonéphrite lupique
La sécurité et l’efficacité de Benlysta par voie intraveineuse chez les enfants et adolescents de moins de 18 ans n’ont pas été évaluées. Aucune donnée n’est disponible.
Mode d’administration
Benlysta est administré par perfusion intraveineuse et doit être reconstitué et dilué avant son administration. Pour les instructions de reconstitution, de dilution et de conservation du médicament avant son administration, voir rubrique 6.6.
Benlysta doit être administré par perfusion pendant 1 heure.
Benlysta ne doit pas être administré par injection intraveineuse en bolus.
Le débit de la perfusion peut être ralenti ou la perfusion interrompue si le patient développe une réaction à la perfusion. La perfusion doit être immédiatement interrompue si le patient présente un effet indésirable susceptible d’engager le pronostic vital (voir rubriques 4.4 et 4.8).
4.3 Contre-indications
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.8 Effets indésirables
Résumé du profil de sécurité chez les adultes
La sécurité du bélimumab chez les patients présentant un lupus systémique a été évaluée au cours de trois études contrôlées versus placebo par voie intraveineuse réalisées avant l’autorisation de mise sur le marché de Benlysta, ultérieurement dans une étude contrôlée versus placebo par voie intraveineuse, dans une étude contrôlée versus placebo par voie sous-cutanée et dans deux études contrôlées versus placebo par voie intraveineuse menées après commercialisation ; la sécurité chez les patients atteints de glomérulonéphrite lupique a été évaluée dans une étude par voie intraveineuse contrôlée versus placebo.
Les données présentées dans le tableau ci-dessous reflètent, chez 674 patients atteints de LS de trois études cliniques réalisées avant l’autorisation de mise sur le marché et chez 470 patients de l’étude ultérieure contrôlée versus placebo, une exposition à Benlysta administré par voie intraveineuse (10 mg/kg de poids corporel administré sur une période d’une heure aux jours 0, 14, 28 puis tous les 28 jours pendant au maximum 52 semaines), et chez 556 patients atteints de LS, une exposition à Benlysta administré par voie sous-cutanée (200 mg une fois par semaine durant au maximum 52 semaines). Les données de sécurité présentées couvrent des périodes pouvant dépasser 52 semaines chez certains patients atteints de LS. Les données reflètent une exposition supplémentaire chez 224 patients atteints de glomérulonéphrite lupique qui ont reçu Benlysta par voie intraveineuse (10 mg/kg de poids corporel durant au maximum 104 semaines). Les données après commercialisation sont également incluses.
La majorité des patients recevait également un ou plusieurs traitements concomitants pour le lupus systémique comme : corticoïdes, immunosuppresseurs, antipaludéens, anti-inflammatoires non stéroïdiens.
Des effets indésirables ont été rapportés chez 84 % des patients traités par Benlysta et 87 % des patients sous placebo. L’effet indésirable le plus fréquemment rapporté (incidence ≥ 5 % chez les patients présentant un lupus systémique traités par Benlysta conjointement aux traitements standards et supérieure ≥ 1 % par rapport au bras placebo) était : rhinopharyngite. La proportion de patients ayant interrompu le traitement en raison d’effets indésirables était de 7 % pour les patients traités par Benlysta et de 8 % pour ceux sous placebo.
Les effets indésirables les plus fréquemment rapportés (> 5 % des patients atteints de glomérulonéphrite lupique traités par Benlysta associé à un traitement standard) étaient : infection des voies respiratoires supérieures, infection des voies urinaires et zona. La proportion de patients ayant arrêté le traitement en raison d’effets indésirables était de 12,9 % pour les patients traités par Benlysta et de 12,9 % pour ceux sous placebo.
Réactions cutanées sévères : des syndromes de Stevens-Johnson (SSJ) et des nécrolyses épidermiques toxiques (NET) ont été rapportés en association avec Benlysta (voir rubrique 4.4).
Résumé tabulé des effets indésirables
Les effets indésirables sont présentés ci-dessous selon la classification par système d’organe MedDRA et par fréquence. Les catégories de fréquence utilisées sont les suivantes :
Très fréquent > 1/10
Fréquent 1/100 à < 1/10
Peu fréquent 1/1 000 à < 1/100
Rare 1/10 000 à < 1/1 000
Fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles).
Dans chaque groupe de fréquences, les effets indésirables sont présentés par ordre décroissant de gravité.
La fréquence donnée est celle la plus élevée observée entre les deux formulations.
Classe de système d’organe | Fréquence | Effets indésirables |
Infections et infestations1 | Très fréquent | Infections bactériennes, par exemple, bronchite, infection urinaire. |
Fréquent | Gastroentérite virale, pharyngite, rhinopharyngite, infections virales des voies respiratoires supérieures | |
Affections hématologiques et du système lymphatique | Fréquent | Leucopénie |
Affections du système immunitaire | Fréquent | Réactions d’hypersensibilité2 |
Peu fréquent | Réaction anaphylactique | |
Rare | Réactions d'hypersensibilité retardée non aiguës | |
Affections psychiatriques | Fréquent | Dépression |
Peu fréquent | Comportement suicidaire, idées suicidaires | |
Affections du système nerveux | Fréquent | Migraine |
Affections gastro-intestinales | Fréquent | Diarrhée, nausées |
Affections de la peau et du tissu sous-cutané | Fréquent | Réactions au site d’injection3, urticaire, rash |
Peu fréquent | Angiœdème | |
Fréquence indéterminée | Syndrome de Stevens-Johnson, nécrolyse épidermique toxique | |
Affections musculo-squelettiques et systémiques | Fréquent | Douleurs aux extrémités |
Troubles généraux et anomalies au site d’administration | Fréquent | Réactions systémiques liées à la perfusion ou à l’injection2, fièvre |
1 Voir « Description de certains effets indésirables » et rubrique 4.4 « Infections » pour plus d’informations.
2 ‘Réactions d’hypersensibilité’ regroupe un ensemble de termes incluant notamment l’anaphylaxie, et pouvant se manifester par différents symptômes tels que : hypotension, angiœdème, urticaire ou autre éruption cutanée, prurit et dyspnée. ‘Réactions systémiques liées à la perfusion ou à l’injection’ regroupe un ensemble de termes pouvant se manifester par différents symptômes tels que : bradycardie, myalgie, maux de tête, éruption cutanée, urticaire, fièvre, hypotension, hypertension, vertiges et arthralgies.
Du fait d’une similitude des signes et symptômes, il n’est pas toujours possible de différencier les réactions d’hypersensibilité des réactions liées à la perfusion ou des réactions systémiques liées à l’injection.
3 s’applique à la formulation sous-cutanée uniquement.
Description de certains effets indésirables
Les données présentées ci-dessous sont les résultats groupés des trois études avant l’autorisation de mise sur le marché par voie intraveineuse (dose intraveineuse de 10 mg/kg de poids corporel seulement) associés à ceux de l’étude par voie sous-cutanée. Les infections et les troubles psychiatriques incluent également des données d’étude après commercialisation.
Réactions systémiques liées à la perfusion ou à l’injection et réactions d’hypersensibilité : les réactions systémiques liées à la perfusion ou à l’injection et les réactions d’hypersensibilité ont été généralement observées le jour de l’administration du traitement, mais des réactions d’hypersensibilité aiguë peuvent aussi survenir quelques jours après la prise du traitement. Les patients présentant des antécédents d’allergies médicamenteuses multiples ou d’hypersensibilité significative peuvent être exposés à un risque accru de réactions.
L’incidence des réactions liées à la perfusion et des réactions d’hypersensibilité après une administration intraveineuse survenant durant les 3 jours suivants la perfusion a été de 12 % dans le groupe recevant Benlysta et 10 % dans le groupe placebo, et respectivement 1,2 % et 0,3 % ont nécessité un arrêt définitif du traitement.
Infections : l’incidence globale des infections, observée dans les études cliniques de pré-enregistrement dans le LS par voie intraveineuse et sous-cutanée, a été de 63 % dans le groupe recevant Benlysta comme dans le groupe placebo. Les infections survenues chez au moins 3 % des patients recevant Benlysta et avec une incidence supérieure d’au moins 1 % par rapport à l’incidence dans le bras placebo ont été : infections virales des voies respiratoires supérieures, bronchites et infections urinaires bactériennes. Cinq pour cent des patients recevant Benlysta ou du placebo ont eu des infections graves ; des infections opportunistes graves ont été diagnostiquées pour respectivement 0,4 % et 0 % de ces patients. Des infections entraînant l’arrêt définitif du traitement sont survenues chez 0,7 % des patients recevant Benlysta et 1,5 % de ceux recevant le placebo. Certaines infections étaient sévères ou d’issue fatale.
Pour plus d’informations sur les infections observées chez les patients pédiatriques dans le LS, se référer à la section Population pédiatrique ci-dessous.
Dans l’étude sur la glomérulonéphrite lupique, les patients recevaient un traitement de fond standard (voir rubrique 5.1) et l’incidence globale des infections était de 82 % dans le groupe recevant Benlysta, contre 76 % dans le groupe placebo. Des infections graves sont survenues chez 13,8 % des patients recevant Benlysta et chez 17,0 % des patients recevant du placebo. Des infections d’issue fatale sont survenues chez 0,9 % (2/224) des patients recevant Benlysta et chez 0,9 % (2/224) des patients recevant le placebo.
Dans une étude post-commercialisation de sécurité de 52 semaines dans le LS, randomisée (1: 1), en double aveugle, contrôlée versus placebo (BEL115467), qui a évalué la mortalité et des événements indésirables d’intérêt particulier chez l'adulte, des infections graves sont survenues chez 3,7 % des patients recevant Benlysta (10 mg/kg de poids corporel par voie intraveineuse) et chez 4,1 % des patients recevant le placebo. Des infections avec issue fatale (par exemple pneumonie et septicémie) sont survenues chez 0,45 % (9/2002) des patients recevant Benlysta et chez 0,15 % (3/2001) des patients recevant le placebo, tandis que la fréquence de la mortalité toutes causes confondues était de 0,50 % (10/2002) chez les patients recevant Benlysta et 0,40 % (8/2001) chez les patients recevant le placebo. La plupart des infections avec issue fatale ont été observées au cours des 20 premières semaines de traitement par Benlysta.
Troubles psychiatriques : dans les études cliniques réalisées dans le LS avant l’autorisation de Benlysta, administré par voie intraveineuse, des événements psychiatriques graves ont été rapportés chez 1,2 % (8/674) des patients recevant Benlysta 10 mg/kg de poids corporel et chez 0,4 % (3/675) des patients sous placebo. Des dépressions graves ont été rapportées chez 0,6 % (4/674) des patients recevant Benlysta 10 mg/kg de poids corporel et chez 0,3 % (2/675) des patients sous placebo. Deux cas de suicide ont été rapportés chez les patients traités par Benlysta (dont un recevant une dose de Benlysta de 1 mg/kg de poids corporel).
Dans une étude réalisée après commercialisation dans le LS, des troubles psychiatriques graves ont été rapportés chez 1,0 % (20/2002) des patients recevant Benlysta et 0,3 % (6/2001) des patients sous placebo. Des dépressions graves ont été rapportées chez 0,3 % (7/2002) des patients recevant Benlysta et moins de 0,1 % (1/2001) des patients sous placebo. La fréquence globale d’idées ou de comportements suicidaires graves ou d’automutilation sans intention suicidaire étaient de 0,7 % (15/2002) chez les patients recevant Benlysta et de 0,2 % (5/2001) dans le groupe placebo. Aucun suicide n’a été rapporté dans les deux groupes.
Les études ci-dessus par voie intraveineuse dans le LS n’excluaient pas les patients ayant des antécédents de troubles psychiatriques.
Dans l’étude clinique par voie sous-cutanée dans le LS, qui excluait les patients avec des antécédents de troubles psychiatriques, des événements psychiatriques graves ont été rapportés chez 0,2 % (1/556) des patients recevant Benlysta et aucun cas n’a été rapporté chez les patients sous placebo. Aucun événement lié à des dépressions graves ni aucun suicide n’ont été rapportés dans les deux groupes.
Leucopénie : l’incidence de la leucopénie rapportée chez les patients atteints de LS en tant qu’effet indésirable a été de 3 % dans le groupe recevant Benlysta et de 2 % dans celui sous placebo.
Troubles gastro-intestinaux : les patients en surpoids (IMC > 30 kg/m2) atteints de LS traités par Benlysta administré par voie intraveineuse ont présenté plus fréquemment des nausées, vomissements et diarrhées que ceux sous placebo, comparativement aux patients de poids normal (IMC ≥ 18,5 à ≤ 30 kg/m2). Aucun des troubles gastro-intestinaux survenus chez les patients en surpoids n’a été grave.
Population pédiatrique
Le profil des effets indésirables chez les patients pédiatriques est basé sur les données d’une étude avec une administration par voie sous-cutanée et d’une d’étude avec une administration par voie intraveineuse.
Dans une étude ouverte de 52 semaines dans laquelle 25 patients pédiatriques (âgés de 10 à 17 ans) atteints de LS ont reçu Benlysta par voie sous-cutanée à une exposition comparable à celui des adultes (200 mg à intervalles fixes calculés selon le poids), avec des traitements standards concomitants, le profil de sécurité chez les patients pédiatriques recevant Benlysta par voie sous-cutanée était comparable avec le profil de sécurité connu de bélimumab.
Dans une étude contrôlée versus placebo de 52 semaines, dans laquelle 53 patients (âgés de 6 à 17 ans) atteints de lupus systémique ont reçu 10 mg/kg de poids corporel de Benlysta par voie intraveineuse aux jours 0, 14, 28, puis tous les 28 jours avec des traitements standards concomitants. Aucun nouveau signal de sécurité n’a été identifié chez les patients pédiatriques âgés de 12 ans et plus (n=43). Les données de sécurité chez les enfants âgés de moins de 12 ans (n=10) sont limitées.
Infections
Groupe de patients âgés de 5 à 11 ans : des infections ont été rapportées chez 8/10 patients recevant Benlysta par voie intraveineuse et 3/3 patients recevant un placebo, et des infections graves ont été rapportées chez 1/10 patient recevant Benlysta par voie intraveineuse et 2/3 patients recevant un placebo (voir rubrique 4.4).
Groupe de patients âgés de 12 à 17 ans : des infections ont été rapportées chez 22/43 patients recevant Benlysta par voie intraveineuse et 25/37 patients recevant un placebo et des infections graves ont été rapportées chez 3/43 patients recevant Benlysta par voie intraveineuse et 3/37 patients recevant un placebo. Lors de la phase d'extension de l’étude en ouvert, il y a eu une infection d’issue fatale chez un patient recevant Benlysta par voie intraveineuse.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration :
Belgique | Luxembourg |
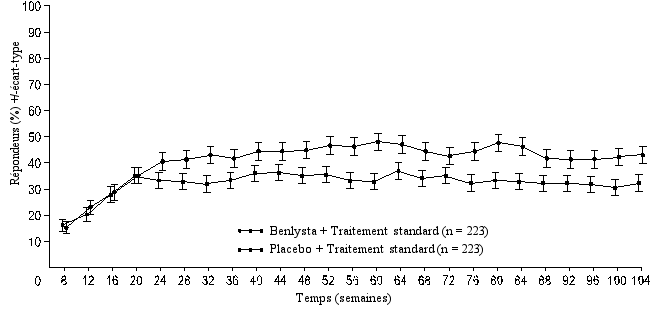
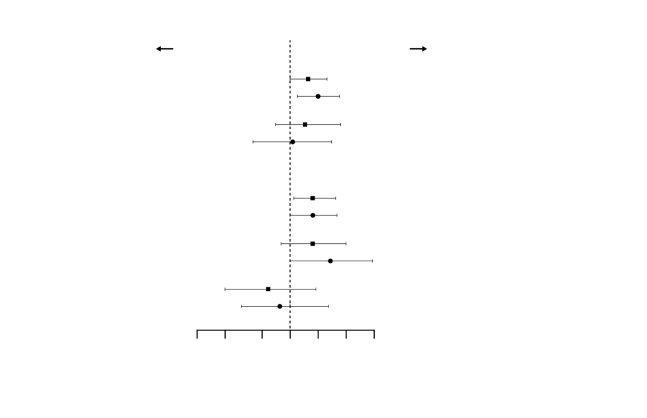
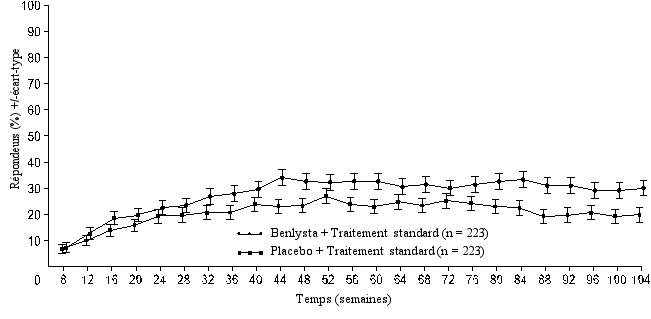 Réponse rénale complète (Complete Renal Response, CRR)
Réponse rénale complète (Complete Renal Response, CRR)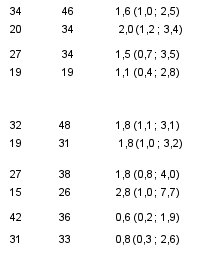


7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHÉ
GlaxoSmithKline (Ireland) Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Irlande
8. NUMÉRO(S) D’AUTORISATION DE MISE SUR LE MARCHÉ
EU/1/11/700/001 1 flacon – 120 mg
EU/1/11/700/002 1 flacon – 400 mg
10. DATE DE MISE À JOUR DU TEXTE
18/07/2025
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’Agence Européenne du Médicament https://www.ema.europa.eu.
PRIX
| Code CNK | Emballage | Code ATC5 | Prix | Prix ex-usine | Sur prescription | Ticket modérateur intervention régulière | Ticket modérateur intervention majorée |
|---|---|---|---|---|---|---|---|
| 2926277 | BENLYSTA PULV SOL DIL PERFUSION 1 FL 400 MG | L04AA26 | - | € 434 | Oui | - | - |
| 2926285 | BENLYSTA PULV SOL DIL PERFUSION 1 FL 120 MG | L04AA26 | - | € 130,2 | Oui | - | - |